Symptoms
Signs of the disease are different, they depend on where exactly the amyloid deposits are localized, how widespread the disease is, and whether there are complications. Often there is a complex of symptoms reflecting damage to several organs.
With amyloidosis of the gastrointestinal tract the following are observed:
- tongue enlargement;
- difficulty swallowing;
- bowel dysfunction;
- heartburn, nausea;
- stomach ache.
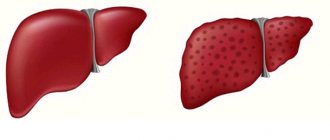
Signs of liver amyloidosis:
- change in liver size;
- pain in the hypochondrium on the right;
- nausea, belching;
- jaundice.
Pancreatic amyloidosis is characterized by dull pain in the left hypochondrium.
Cardiac amyloidosis is expressed in rhythm disturbances, myocardial lesions, and heart failure.
Amyloidosis of the nervous system has the following symptoms:
- peripheral polyneuropathy (numbness of the limbs, tingling, burning sensation);
- headaches, dizziness, increased sweating;
- urinary and fecal incontinence;
- sexual dysfunction.
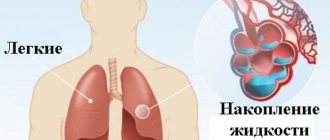
With amyloidosis of the respiratory system, hoarseness of the voice and bronchitis are observed.
A case of primary amyloidosis with predominant involvement of the heart, lungs and pleura
Over the past decades, ideas about amyloidosis have changed radically, which is associated with the establishment of heterogeneity in the protein composition of amyloid fibrils. The modern classification is based on the specificity of the amyloid protein. There are systemic and local forms of amyloidosis. The prevalence of AL amyloidosis, according to the US National Center for Health Statistics, is 4.5 cases per 100,000 [1]. The age-standardized incidence increase is 5.1–12.8 per million person-years, representing approximately 3200 new cases per year in the United States [2]. It is currently believed that the development of primary idiopathic amyloidosis is based on benign plasma cell dyscrasia of the bone marrow. An abnormal clone of plasma cells produces amyloidogenic immunoglobulins, which are light chains of monoclonal immunoglobulin, usually lambda (l), less often kappa type (k) (their ratio is 3:1). Amino acid analysis shows that some amino acids in the variable regions of the light chains of these immunoglobulins occupy an unusual position, which leads to instability and a tendency to fibrillogenesis. It was noted that the most common subgroups of light chains in AL deposits are represented by types VI and I. This suggests that they are considered amyloidogenic. Subsequently, the process of precipitation of amyloid proteins occurs in the tissues of target organs [3]. The main target organs for AL amyloidosis are the heart, kidneys, gastrointestinal tract, respiratory system and other organs. The clinical picture of primary amyloidosis is diverse and is primarily due to the predominant damage to certain organs. The first symptoms of the disease are weakness and weight loss. Damage to the cardiovascular system, according to O.M. Vinogradova (1980), clinically occurs in 70% of patients, and in anatomical studies - in 87% of cases. Heart pathology is the leading manifestation of primary amyloidosis and is primarily characterized by myocardial damage. Amyloid is deposited intermuscularly, perivascularly, causing atrophy of muscle fibers. The myocardium loses its elasticity and becomes rigid, as a result of which the function of diastolic relaxation suffers. The cavities of the left and right ventricles do not dilate, and the cavities of the atria sharply expand. Cardiomegaly develops, up to cor bovinum. The increase in myocardial mass can be so pronounced that at autopsy the heart occupies most of the chest (the mass of the heart can reach up to 1 kg). Amyloid deposits can be located in the conduction system of the heart, which leads to various rhythm disturbances and can cause sudden death [4]. Amyloid deposition also occurs in the walls of the coronary vessels, causing myocardial ischemia. Involvement of the valve apparatus occurs in 12–15% of patients and in 35% of anatomical studies. As a rule, there is thickening and deformation of the valves with the development of their insufficiency, and when deposited in the area of the fibrous ring, stenosis is formed [5]. The most typical signs detected during instrumental examination are low ECG voltage, arrhythmias and conduction disturbances, pathological Q waves simulating myocardial infarction; EchoCG signs: symmetrical thickening of the ventricular walls, a picture of restrictive cardiomyopathy with signs of diastolic dysfunction; Many authors describe echo-positive inclusions in the myocardium in the form of small granules, which are considered to be particles of amyloid inclusions; pericardial effusion is possible [6]. Damage to the respiratory system in systemic forms of amyloidosis is quite common. So, according to O.M. Vinogradova (1980), clinical signs of damage to the respiratory system were observed in 50% of patients and in 83% according to pathological data. There are diffuse and local lung damage. Weis (1960) in his classification distinguished generalized amyloidosis, subdivided into nodular and diffuse pulmonary and limited amyloidosis, in which tracheobronchial and isolated nodular pulmonary amyloidosis were separately distinguished. Subsequently, mixed diffuse-nodular forms of amyloidosis were described). John L. Berk identifies 5 forms of extravascular lung damage in AL amyloidosis [7]: 1. Diffuse interstitial, or alveolar-septal damage, in which amyloid is deposited between the vascular endothelium and the alveolar epithelium of the pulmonary interstitium. 2. Nodular deposition. 3. Damage to the pleura. 4. Intra- and extrathoracic adenopathy. 5. Damage to the diaphragm, which is extremely rare. The diffuse-interstitial form deserves special attention, since it is it that is combined with the cardiopathic variant of the course of primary amyloidosis and is often not diagnosed, since its clinical and radiological picture is mistaken for manifestations of congestive heart failure. John L. Berk [7] presented autopsy data on 12 AL patients: all had deposits in the interstitium, alveolar septa, basement membranes between alveolar epithelial cells and capillary endothelial cells, walls of small vessels and airways. Despite the widespread histological appearance of amyloid lesions, 64% of patients had no clinical signs of airway involvement. Only in 1 case out of 12 did lung damage determine the outcome of the disease. According to other authors, diffuse interstitial damage was the cause of death in 10% of patients. Clinical signs of this variant are progressive shortness of breath and dry paroxysmal cough. When examining the function of external respiration, a restrictive type of ventilation disorders is revealed. X-ray and CT examinations reveal a diffuse enhancement of the pulmonary pattern due to the vascular and interstitial component. Damage to the pleura in AL amyloidosis is rare and is observed in approximately 6% of patients (according to O.M. Vinogradova, it is 6.6%). John L. Berk indicates 6% [8], characterized by the formation of pleurisy of an aggressive course and torpid to the diuretic therapy. Pleural effusion can be unilateral, but more often it is bilateral. It is predominantly transudative in nature, although there are also exudates. Lymphocytosis predominates in the cytogram. Pleural effusion is very common in patients with amyloid cardiomyopathy, so the development of pleural effusion is associated with decompensated heart failure. However, a study conducted by the Boston University Medical Center, which included 636 patients during the period 1994–2001, refutes this opinion [8]. Two groups of patients with AL amyloidosis were analyzed: the first included patients with amyloid heart disease and pleural effusion (35 people), the second group with amyloid cardiomyopathy without pleural effusion (120 people) [8]. The results of the study showed that left ventricular dysfunction and low plasma oncotic pressure are not fundamental in the development of pleural effusion. The formation of pleural effusion is associated primarily with the deposition of amyloid in the intercellular spaces, in the submesothelial lymphatic vessels. Amyloid deposits inhibit the resorption of pleural fluid, as a result of which the drainage ability of the pleura is impaired. In turn, amyloid deposits also change the secretory function of the pleura [9]. Diagnosis of amyloidosis is complex and requires a complex of laboratory and instrumental studies, but the final diagnosis must be confirmed morphologically. Changes in laboratory data are nonspecific and reflect dysfunction of a particular affected organ. In 85% of patients, an immunoelectrophoretic study of serum protein and urine reveals a monoclonal paraprotein, which is represented by light chains of immunoglobulins, more often l-chains, less often k-chains. A coagulation study reveals a deficiency of coagulation factor X. Bone marrow examination shows moderate plasmatization of 5–10% (up to 20%). The combination of a characteristic clinical picture with the presence of monoclonal proteins in the blood serum and urine and an increase in plasma cells in the bone marrow is insufficient to establish a diagnosis of primary amyloidosis. A reliable method is histological examination of the affected organ (kidney, liver, myocardium, etc.) [10]. A biopsy of the rectal mucosa is quite informative, in which the probability of detecting amyloid is 50–70%. A biopsy of the gum mucosa is not very informative; aspiration biopsy of subcutaneous fat from the anterior abdominal wall is widely used abroad. The more widespread the process, the higher the likelihood of detecting amyloid in various places. To determine amyloid in histological material, staining methods are used: if congophilia is detected, the preparation is examined in polarized light to identify the effect of birefringence, with only amyloid acquiring an apple-green color. For amyloid typing, a staining method is used with an alkaline guanidine solution, which changes the congophilic properties of various types of amyloid depending on the exposure time in the guanidine solution. But the most accurate is an immunohistochemical study using monoclonal antibodies to amyloid precursor proteins. Diagnosis of amyloidosis is a difficult task, which is confirmed by the observation we presented.
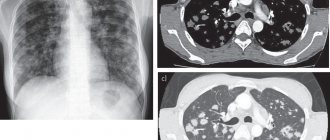
Patient K., 54 years old, was admitted to the clinic with complaints of shortness of breath with slight physical exertion, a dry cough that occurs when changing body position, pressing pain in the chest during physical activity and at rest, severe general weakness, lack of appetite, weight loss over the past year by 16 kg. From the anamnesis it is known that she first began to notice an increase in blood pressure during her first pregnancy at the age of 23, and nephropathy of pregnancy was diagnosed. From the age of 30, blood pressure levels stabilized at 140–150/90–100 mm Hg. with an increase to 170–190/90–100 mm Hg. She did not receive constant antihypertensive therapy; she took clonidine situationally. In 1986, kidney scintigraphy was performed, which revealed a slight decrease in excretory function on both sides, without changes in secretory function. An ultrasound examination revealed bilateral pyelectasis. Leukocyturia was observed in urine tests. Based on this, it was concluded that the patient had chronic pyelonephritis and the formation of secondary arterial hypertension. Since 1998, the patient began to notice the appearance of hemorrhagic rashes the size of a match head on the skin of the paraorbital areas, which became hyperpigmented over time. In 2000, persistent hoarseness appeared against the background of ARVI. In 2001, after undergoing supravaginal amputation of the uterus with left appendages due to uterine fibroids and endometriosis of the left ovary, the course of arterial hypertension was complicated by high rates, the patient began to notice the appearance of shortness of breath during normal physical activity. EchoCG revealed for the first time LV myocardial hypertrophy (the thickness of the interventricular septum and the thickness of the posterior wall was 12 mm) without dilatation of the heart chambers. Enalapril was prescribed, which she took irregularly. After stress in January 2003, mild pressing pain in the chest appeared, decreased tolerance to physical activity in the form of shortness of breath, and weakness. I didn’t go to the doctors. In the summer, she noted the appearance of edema in the lower extremities and the progression of shortness of breath. In September, after physical overexertion for a week, she noted a sharp deterioration in her health: increased shortness of breath, the appearance of intense pain in the chest, increased swelling of the legs and feet. When visiting the clinic, an ECG revealed cicatricial changes in the anteroseptal region and the apex of the LV; echoCG revealed akinesis of the anteroseptal region and a decrease in the LV ejection fraction; According to chest x-ray, congestion in the lungs, bilateral hydrothorax. The patient was urgently hospitalized in ICU 3 of the Central Military Hospital named after. A.V. Vishnevsky, where no data in favor of acute myocardial infarction were obtained. The condition was regarded as post-infarction cardiosclerosis. During therapy with nitrates, ACE inhibitors and diuretics, the condition improved (swelling in the legs disappeared, shortness of breath decreased, and at the time of discharge there was no fluid in the pleural cavities). She refused the proposed coronary angiography. Subsequently, the symptoms of heart failure increase again, and a tendency toward normotension appears. In June 2004, he was hospitalized at the All-Russian Scientific Research Center, where echocardiography showed a picture of left ventricular myocardial hypertrophy (TZS-TMZH - 14 mm), expansion of the left ventricular cavity to 4.2 cm, impaired diastolic function of the left ventricular myocardium, EF - 60%, signs of pulmonary hypertension were revealed for the first time (MPAP 45 mm Hg), initial symptoms of stenosis of the left AV orifice, mitral regurgitation grade 2. Coronary angiography did not reveal signs of stenosis of the coronary arteries. The condition was regarded as the formation of a hypertensive heart in a patient with long-term uncorrectable arterial hypertension, changes on the ECG in the form of scars and the presence of angina syndrome were regarded as manifestations of hypertrophic cardiomyopathy. Therapy with bisoprolol, enalapril, and hydrochlorothiazide was prescribed. Blood pressure levels returned to normal, but shortness of breath and weakness persisted. Deterioration in condition since February 2005. Shortness of breath is increasing again, which required increased diuretic therapy, and a tendency to hypotension has appeared. In April 2005, dryness and an unpleasant taste appeared in the mouth, loss of appetite, nagging pain in the epigastrium, chills after eating, and the patient began to lose weight. Endoscopy revealed no pathology; ultrasound of the abdominal cavity revealed signs of diffuse changes in the pancreas, bilateral hydrothorax. In June, a dry hacking cough appears, shortness of breath and weakness progress. In October, the patient was hospitalized in the hospital, by this time the weight loss was 10 kg, which forced an oncological search (CT of the abdominal cavity, CT of the pelvis, ultrasound of the mammary glands, thyroid gland, genitals, endoscopy, colonoscopy), which did not reveal oncopathology. CT examination of the chest organs revealed: right-sided pleural effusion, a zone of compaction of lung tissue with a diameter of 10 mm in the X segment of the right lung, enrichment of the vascular pattern due to thickening of the interlobular septa. These changes were interpreted as right lower lobe pneumonia, right pleural effusion, interstitial edema, or lymphogenous carcinomatosis. A pleural puncture was performed; the examination revealed transudate, but no atypical cells were found. In biochemical (electrophoresis of protein fractions was not performed) and immunological blood tests - no pathological changes, with the exception of a 10-fold increase in the level of ovarian oncogene (CA 125). Antibacterial, antihypertensive (perindopril, nebivolol) and diuretic (hydrochlorothiazide 100 mg) therapy was administered, and with slight improvement the patient was discharged home. The patient's condition is slowly deteriorating, shortness of breath progresses, fluid accumulates in the pleural cavities, nocturnal orthopnea occurs without an increase in blood pressure, which forces us to continue the diagnostic search. Repeated CT examinations of the lungs reveal an increase in the pulmonary pattern, which is regarded as interstitial and incipient alveolar pulmonary edema. The patient is admitted to the pulmonology department of the I.M. MMA hospital therapy clinic. Sechenov. On admission: condition of moderate severity. Asthenic physique, low nutrition. Weight 50 kg, height – 162 cm. BMI – 19. The skin is pale and dry. Cyanosis of lips. The axillary lymph nodes are palpated. Varicose veins of the superficial veins of the lower extremities. The lower legs are pasty, more so on the right one (due to venous insufficiency). Nasal breathing is free. NPV – 20 per 1 min. at rest. The chest is of normal shape, the right half lags behind in the act of breathing. In the lower parts of both lungs, percussion revealed dullness to the level of the 5th rib. Breathing over the lungs is vesicular with a hard tint, breathing is not carried out on the right in the basal sections, and weakened on the left. The heart area is not changed. The boundaries of relative dullness of the heart are expanded to the left. Heart sounds are muffled, rhythmic, heart rate – 80 beats. per minute The emphasis of the 2nd tone is on the aorta and pulmonary artery; a systolic murmur is heard at the apex. Blood pressure – 90/60 mm Hg. The abdomen is soft on palpation, sensitive in the epigastrium, and painless in other parts. The liver is lowered, the edge is smooth, painless. The spleen is not enlarged. The kidneys are not palpable. The symptom of effleurage is negative. There is no dysuria. There are no gross neurological symptoms. Blood tests: ESR – 3 mm/h, leukocytes – 11000, neutrophils – 74%, eosinophils – 4%, monocytes – 2%, lymphocytes – 16%, basophils – 1%. Hemoglobin – 145 g/l, erythrocytes – 4.7 million, platelets – 443,000, CP – 0.92. total protein – 7 (6–8) g/dl, albumin – 4.1 g/dl, creatinine – 1.1 mg/dl. An electrophoretic study of protein fractions revealed an M gradient in the gamma globulin zone; an immunochemical study of serum proteins revealed paraproteinemia G, which amounted to 12.7% of the total serum protein, or 9.1 g/l, the level of beta-2 microglobulin and CRP are normal. Increase in the level of tumor marker CA 125 by 3 times. A general urinalysis revealed trace proteinuria, otherwise without pathological abnormalities. An electrophoretic study of urine proteins did not reveal Bence-Jones protein (including immunofixation with antiserum to free light chains). An X-ray of the chest organs revealed fluid in the pleural cavities on the right up to the 6th rib, and on the left – the 6th intercostal space. In the lower lobes of both lungs, a diffuse deformation of the pulmonary pattern was determined, having a mesh-cellular structure; the vascular pattern was not traced in these zones. The apices of the lungs are not changed. The roots of the lungs are not expanded, structural, the width of the pulmonary artery at the level of the right lung is 1.3 cm (normal). The heart is horizontal. The aorta is dilated and lengthened (Fig. 1). CT scan of the lungs reveals fluid in the pleural cavities, more in the right one. The pulmonary pattern is enhanced in all sections due to the vascular component. There was a slight enlargement of the mediastinal lymph nodes. EchoCG revealed concentric hypertrophy of the LV myocardium (TMZH-TZS – 14 mm) with dilatation of the LA cavity to 4.8 cm, LV EDR/ESR, respectively, 4.9/3.6 cm, EF – 52%, RV dimensions – 2.7 cm, moderate hypertrophy of the RV wall – 0.52 cm. Slight decrease in global LV systolic function. Fibrosis of the MV leaflets, an increase in the pressure gradient on the MV to 8 mm Hg, cross-sectional area of 3.7 cm, but the movement of the MV leaflets is in antiphase. MV regurgitation grade 2–3. Type 2 LV diastolic dysfunction. The average pressure in the LA is 20 mm Hg. Art. (at normal – up to 15 mm Hg). A small amount of fluid in the pericardial cavity. A specific opalescent, enhanced echo signal from the walls of the left ventricle was noteworthy. For the first time, cardiac amyloidosis was suggested. FVD - significant ventilation disorders of a restrictive type (VC - 1.93 l (66%), FEV1 - 1.53 l (62%). The patient underwent puncture of the right pleural cavity, 500 ml of straw-yellow transparent fluid was evacuated. During the study specific gravity 1003, protein 5.05%, glucose 150 mg%, Rivalta test - negative, cytological examination - leukocytes - 5-10-15, lymphocytes - 75%, neutrophils - 25%, erythrocytes - 40-60. Atypical cells and CD were not found. PCR and luminescent microscopy studies did not reveal Mycobacterium tuberculosis. In the sternal puncture there was a slight increase in plasma cells (up to 1.8%). Probable diagnoses were discussed: Meigs syndrome, pleural mesothelioma, pulmonary lymphongitis, tuberculosis. Pelvic ultrasound performed did not reveal any pathology in the right ovary, blocks of biopsy specimens of the uterus and left ovary removed in 2001 were reviewed - no data confirming the neoplastic process was obtained.Thus, the diagnosis of Meigs syndrome became extremely doubtful. The absence of Mycobacterium tuberculosis in the study of pleural fluid, specific lesions of the lungs and foci outside pulmonary tuberculosis made it possible to exclude the tuberculous nature of the disease. To clarify the diagnosis, videothoracoscopy with atypical resection of the upper lobe of the right lung was performed. During the operation, 1.5 liters of clear yellow liquid was aspirated. The parietal and visceral pleura are not thickened, the vessels are injected. Congestion of the lung was noted. In the 1st segment there is an area of scarring of the lung tissue with a thickened whitish pleura. Mechanical and chemical pleurodesis was performed using a 1% iodopyrone solution. Morphological examination revealed lung tissue with pleural fibrosis. In the fibrotic pleura there are focal lymphohistiocytic infiltrates, vascular sclerosis and, in places, lymphostasis and hemorrhage. In the subpleural sections there is interstitial fibrosis and hyalinosis, in the lumen of individual alveoli there are large macrophages with brown pigment in the cytoplasm. When staining with Congo-mouth and thioflavin, amyloid deposition was detected in the walls of blood vessels, the pulmonary interstitium and in the pleura. Typing confirmed the presence of AL amyloidosis. Genetic testing of blood serum did not reveal mutations in the transthyretin gene. After the diagnosis was established, the patient refused treatment. Subsequently, the patient was hospitalized in a medical institution in Moscow, where, in her words, a course of therapy with melphalan and prednisolone was administered in gentle doses. The second course was not carried out due to poor tolerability of treatment. After 4 months the patient died due to progressive symptoms of cardiovascular failure. Thus, the patient was diagnosed with AL amyloidosis with damage to the lungs and pleura (diffuse interstitial type), heart (circulatory failure stage II A with impaired diastolic function of a restrictive type, damage to the mitral valve with the formation of stenosis of the left AV orifice), autonomic nervous systems (orthostatic hypotension, hoarseness), blood vessels. Upon retrospective analysis of the patient's medical history, it can be stated that the disease debuted in 1998 with skin manifestations - the appearance of petechial rashes in the paraorbital areas. The appearance of hoarseness in 2000 was the first sign of autonomic disorders, which was mistakenly regarded by the patient and doctors as a complication after suffering from the flu. Subsequently, hypertrophic cardiomyopathy is formed, which was considered in the program of uncorrectable arterial hypertension. A very interesting fact is that the formation of mitral stenosis was initially discussed in the program of rheumatic lesions; after rheumatism was rejected, they settled on atherosclerotic origin, despite the fact that the coronary vessels (according to coronary angiography) were intact. Now we can say with confidence that the damage to the fibrous ring of the left AV orifice and the MV leaflets is caused by the deposition of amyloid in them. The first signs of lung damage appeared in 2003; then they were incorrectly interpreted as manifestations of heart failure. The accumulation of transudate in the pleural cavities with preserved myocardial contractility and minor signs of circulatory failure (limited only to the small circle) indicated damage to the pleura and loss of its barrier function. And the development of persistent hypotension in a patient suffering from arterial hypertension for a long time was associated with damage to the autonomic nervous system, and not with progressive heart failure. It should be noted that the occurrence of petechial rashes in the paraorbital areas is a symptom specific to AL amyloidosis and is considered as a pathognomonic and prognostically unfavorable sign of the clinical course of amyloidosis, but, contrary to generally accepted opinion, in the case we presented, 8 (!) passed from the onset of the disease to the final stage. years. Perhaps a correct assessment of this symptom at the onset of the disease would have made it possible to establish a diagnosis at an earlier stage and give more chances for treatment and prolongation of the patient’s life. It should be noted that a feature of this clinical case is the absence of kidney damage even in the advanced stage of the disease, which significantly complicated approaches to diagnosis. Only the combination of seemingly diverse symptoms made it possible to approach the diagnosis of primary amyloidosis, which was confirmed morphologically. In our message, we would like to draw attention to a pathology that is quite rare in both pulmonological and general therapeutic practice. The difficulty of diagnosis, as already noted, is associated with the variety of manifestations of primary amyloidosis. When using generally accepted standards of differential search, the principle of an individual approach to the patient is lost, which makes it difficult to determine the true diagnosis. Studying the developed and recommended differential search schemes for pleural effusions by domestic authors, it should be noted that amyloidosis is not included in this range of diseases. Meanwhile, in articles by foreign authors on this topic, amyloidosis is included in the search for the causes of pleural effusion [11].
References 1. Gertz MA, Kyle RA, Thibodeau SN Familial amyloidosis: a study of 52 North American–born patients examined during a 30–year period. Mayo Clin Proc 1992; 67:428–440. 2. Simms RW, Prout MN, Cohen AS The epidemiology of AL and AA amyloidosis. Bailliers Clin Rheumatol 1994;8:627–634. 3. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995;32:45–59. 4. Koyama J, Ray-Sequin PA, Davidoff R, et al. Usefulness of pulsed tissue Doppler imaging for evaluating systolic and diastolic left ventricular function in patients with AL (primary) amyloidosis. Am J Cardiol 2002; 89:1067–1071. 5. Vinogradova O.M. Primary and genetic variants of amyloidosis. M., “Medicine”, 1980. 6. Moiseev S.V. Infiltrative myocardial lesions. Restrictive cardiomyopathy. M., Pharma Press, 1998. 7. Berk JL, O'Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respirat Crit Care Med 2002; 23:155–165. 8. Berk JL, Keane J, Seldin DC, et al. Persistent pleural effusions in primary systemic amyloidosis. Chest 2003; 124;969–977. 9. Mathews V, Vasudevan AR, Nair S, et al. Primary systemicamyloidosis: pleural involvement with exudative pleural effusion. J Assoc Physicians India 1996; 44:345–346 10. Kozlovskaya L.V., Varshavsky V.A., Chegaeva T.V. and others. Amyloidosis is a modern view of the problem. //Practical nephrology. - 1998. - 2:24–26. 11. Pleural effusion: Article bu Fredrick M Abrahamian, DO Jan.11.2005.
Treatment and prevention
In most cases, amyloidosis is treated at home. If there are complications, the patient may be indicated for hospitalization.
Treatment for amyloidosis includes taking medications and following a number of doctor’s recommendations. But in severe cases, the spleen is removed and kidney or liver transplantation may be required.
The list of medications depends on the location of the deposits, the degree of damage to the body, and existing complications. Thus, with secondary amyloidosis, specific treatment of the primary disease is necessary. In addition, medications are prescribed to eliminate symptoms.
Also, the patient is often prescribed a special diet (restriction of protein and salt intake).
There is no specific preventive program for amyloidosis, since the exact causes of the disease are unknown.
Amyloidosis is a complex disease that requires ongoing treatment. A patient with amyloidosis should be regularly monitored by a specialist and undergo examinations to monitor his health. Medical has modern diagnostic equipment, which allows you to make an accurate diagnosis in the shortest possible time. And the center’s experienced, highly qualified specialists will prescribe effective treatment and ensure proper care for the patient.
Classification (types) of amyloidosis
The Nomenclature Committee of the International Union of Immunological Societies in 1993 developed a classification according to which systemic amyloidosis is divided into:
- secondary to chronic inflammatory diseases or familial Mediterranean fever
- primary, associated with multiple myeloma
- in patients undergoing elective hemodialysis
- hereditary family
Elderly people often develop a localized form of the disease:
- senile A. atrium
- for Alzheimer's disease
- for non-insulin-dependent diabetes mellitus
Diagnostics
Diagnosis of amyloidosis is carried out by laboratory tests and other special studies of human organs.
When recognizing amyloidosis, laboratory tests reveal anemia in the blood, leukocytosis, increased ESR (erythrocyte sedimentation rate), hypoproteinemia and hyperglobulinemia, hyponatremia, hypoprothrombinemia and hypocalcemia. When the liver is damaged, hypercholesterolemia and sometimes hyperbilirubinemia appear. The functions of the thyroid gland and kidneys are assessed. Urine is examined, revealing not only protein in the sediment, but also leukocytes and erythrocytes, casts. Increased amyloid content in urine and blood determines primary amyloidosis. When signs of inflammatory diseases are detected, secondary amyloidosis appears. A coprological study is carried out to identify steatorrhea, amilorrhea, and creatorrhoea.
Other studies to detect amyloidosis are echocardiography, x-ray, in which:
- the esophagus is hypotonic, and peristalsis is weakened; when the patient is in a horizontal position, the barium suspension lingers in the esophagus for a long time - this is esophageal amyloidosis;
- evacuation of contents from the stomach, smoothed folds of the mucous membrane, weakened peristalsis - gastric amyloidosis;
- There is a smoothing of the relief of the intestinal mucosa and expansion of the intestinal loops, thickening of the folds - intestinal amyloidosis.
A biopsy is also performed.
When the gastrointestinal tract is affected, gastric ulcers and chronic gastritis are observed.
Carpal tunnel syndrome and peripheral polyneuropathy, restrictive cardiomyopathy, nephrotic syndrome, urinary tract obstruction, acute tubular necrosis, symmetric polyarthritis, pulmonary disease, kidney toxicity, Alzheimer's disease and dementia with multiple cerebral infarctions lead to amyloidosis.