Primary biliary cirrhosis is a destructive and inflammatory liver disease characterized by a chronic course. It is accompanied by damage to the intrahepatic ducts. As such cirrhosis progresses, ductopenia (disappearing bile duct syndrome) and persistent cholestasis (decreased bile flow into the duodenum due to disturbances in its production, excretion and/or excretion) may develop. In the terminal stage of the disease, liver failure is usually observed.
general description
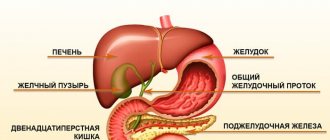
Biliary cirrhosis is
a chronic autoimmune disease that occurs as a result of a violation of the outflow of bile through the intrahepatic and biliary tract (cholestasis) and is characterized by the replacement of parenchymal liver tissue with connective tissue (fibrosis). The prognosis of the disease itself depends on diagnosis and in undiagnosed cases leads to deterioration of development: progressive destruction of the parenchyma with the formation of foci of fibrosis, which results in liver cirrhosis and liver failure.
According to statistics, in economically developed countries, biliary cirrhosis is diagnosed in people aged 30 to 55 years, more often in men. The ratio of incidence among men to women is approximately 3 to 1.
Back
Primary biliary cirrhosis
Primary biliary cirrhosis (PBC) is a chronic granulomatous destructive inflammatory disease of the interlobular and septal bile ducts of an autoimmune nature, leading to the development of prolonged cholestasis, and in later stages to the formation of cirrhosis.
Etiology and pathogenesis
The etiology of PBC is unknown. Genetic factors play a role. Cases of family diseases have been described, but their frequency is low - 17%.
Autoimmune cellular reactions play a leading role in the pathogenesis of PBC. Autoimmune liver diseases are characterized by the presence of specific autoantibodies. PBC is characterized by the presence of antimitochondrial antibodies (AMA), specific for 2-oxoacid dehydrogenase complexes located on the inner membrane of mitochondria. Most often (95–100%) in PBC, autoantibodies to the E2 component of the pyruvate dehydrogenase complex (PDCE2) are detected.
For a long time it was believed that the presence of AMA was only a concomitant feature, but after Gershwin and Mackay discovered the autoantigen, elegantly conducted studies revealed the specificity of the action of AMA, and their role in the pathogenesis of the disease was revealed. These antibodies suppress the activity of PDCE2, which acts as an immunodominant target. AMAs are IgG3 and IgM and are found in the serum and bile of patients. The corresponding epitons of B cells are described. No correlation was found between the amount of AMA and the stage of the disease, but a relationship was shown between the activity of the process and the level of PBC-specific B cells in the blood serum.
The central target for the development of the inflammatory reaction and immune response are the bile ducts. AMAs bind to the apical membrane of bile duct epithelial cells, on the surface of which are proteins of the major histocompatibility complex (MHC) class II. It can be assumed that the pathological expression of the autoantigen occurs before the formation of an immune response with the expression of class II proteins on the cell surface. Further expression occurs in the later stages of disease development; the presence of activated T cells is associated with the ongoing necroinflammatory process in the bile ducts. It is important to note that adhesion molecules that enhance the immune response are found on biliary epithelial cells and lymphocytes.
T lymphocytes play the main role in direct damage to the intrahepatic bile ducts
. In the liver and peripheral blood of patients, CD4-positive PDCE2-specific Thelper cells of both Th1 and Th2 populations are detected. There is evidence that Th1 cells predominate in the liver of patients with PBC; they stimulate the cellular immune response through the production of IL2 and INFg.
The answer to the question of how PDCE2, which are peptides of the body itself, can cause an immune response is provided by the theory of molecular mimicry.
The main mechanism of death of biliary epithelial cells is apoptosis, which is carried out both by Th1, which carries the Fas ligand, and by cytokines secreted by this cell subpopulation [1].
Morphological characteristics
Currently, a classification has been adopted, according to which there are 4 histological stages of PBC: chronic non-purulent destructive cholangitis, ductal stage
;
proliferation of bile ducts and periductal fibrosis ductular stage
;
stromal fibrosis in the presence of inflammatory infiltration of the liver parenchyma;
cirrhosis of the liver .
Chronic non-purulent destructive cholangitis (stage 1) is characterized by inflammation and destruction of predominantly interlobular and septal bile ducts. Dilated portal tracts are infiltrated with lymphocytes, plasma cells, macrophages and eosinophilic leukocytes. Among the cells in the infiltrates of the portal tracts, formed lymphoid follicles are found. The infiltrate of the portal tracts does not extend into the parenchyma; individual lymphocytes or groups of cells can penetrate shallowly into the lobules. Infiltrates are found in the walls of some intralobular bile ducts.
The integrity of the basement membrane of the affected bile ducts is compromised.
Granulomas and granulomatous cholangitis are often found near the affected bile ducts. Granulomas are built from epithelioid and giant multinucleated cells and in most cases are clearly visible in preparations.
Histological signs of cholestasis are usually not detected at this stage.
Proliferation of cholangioles and periductal fibrosis (stage 2). In the portal tracts, along with lymphoplasmacytic infiltration and collapsing bile ducts, foci of proliferation of the biliary epithelium appear. Proliferating cholangioles with infiltrate cells spread into the periportal sections of the lobules. The number of interlobular and septal bile ducts decreases as they are destroyed. A characteristic diagnostic sign of PBC appears: empty portal tracts, the inflammatory infiltrates of which do not contain bile ducts.
Stromal fibrosis in the presence of inflammatory infiltration of the liver parenchyma (stage 3) is characterized by the appearance of connective tissue strands extending from the portal tracts and connecting neighboring tracts (portoportal septa) and central veins with portal tracts (portocentral septa). Along them, the inflammatory infiltrate spreads into the proliferating bile ducts, and the proliferation of the ducts decreases. The reduction of the interlobular and septal bile ducts progresses. This leads to increased cholestasis. The copper content in liver biopsies increases many times over.
Cellular infiltration of the parenchyma and necrosis of hepatocytes intensify, fibrosis increases in the portal tracts, and monolobular false lobules are formed.
Liver cirrhosis (stage 4) is characterized by all the signs of monolobular cirrhosis.
Clinical picture
The disease occurs predominantly in women, most often over the age of 35 years. A distinctive feature of PBC is the relatively rare incidence of men (10-15% of the total incidence of PBC).
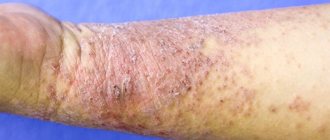
Skin itching is the most characteristic initial symptom of PBC, observed in most patients. Itching of the skin is combined with icteric discoloration of the skin and sclera, but it often precedes jaundice, sometimes by several months or even years. A number of patients we observed over a period of 26 years developed only mild yellowness of the sclera without staining of the skin.
Jaundice of the cholestatic type, slowly increasing, is detected as an early symptom of the disease in less than half of patients. Jaundice, which appears at the time of diagnosis and rapidly increases, can be considered as a prognostically unfavorable symptom, indicating rapid progression of the disease.
Xanthelasma in the early stages is detected in 20-30% of patients. Their formation directly depends on the level and duration of hypercholesterolemia. Extrahepatic signs
“hepatic” palms, spider veins are present only in some patients; they are always singular. Most of the observed men had gynecomastia.
Hepatomegaly is usually minor and is detected in most patients. Splenomegaly is observed in less than half of the patients and is not combined with symptoms of hypersplenism. In the early stages, bone demineralization manifests itself as pain in the lower back, ribs, and joints.
The initial signs of the disease can be nonspecific symptoms such as pain in the right hypochondrium, in some cases with fever; increased ESR; pain in joints and muscles, as well as dyspeptic, skin syndromes, vasculitis, scleroderma. In 20% of patients in the initial stages, the disease can occur without clinical symptoms, while ALP is often elevated, AMA in a titer of 1:40 or higher is always detected, and changes characteristic of PBC are found in liver biopsies.
Advanced stages of PBC are characterized by a progressive deterioration in the condition of patients, an increase in jaundice, sometimes an increase in temperature to subfebrile and then febrile levels, exhaustion (up to cachexia) due to impaired absorption in the intestine. Itching of the skin in the terminal stage of the disease weakens in a number of patients, and disappears with progressive hepatic cell failure.
With the progression of cholestasis, steatorrhea, osteoporosis, and then osteomalacia, xerophthalmia and hemorrhagic syndrome are observed. Fragility of the vertebral bodies, kyphosis and pathological fractures appear. Signs of portal hypertension develop, in particular, varicose veins of the esophagus and stomach. Patients die due to symptoms of hepatic cell failure, which can be provoked by complications of biliary cirrhosis: bone fractures, portal hypertension, ulcerative bleeding.
Late complications of PBC include the development of cholangiocarcinoma, which is much more common in men than in women. The formation of gallstones is also possible.
Systemic manifestations
For biliary cirrhosis, a systemic pattern of lesions is natural, most clearly manifested by changes in the exocrine glands: lacrimal, salivary, pancreas, as well as kidneys (tubulointerstitial nephritis, glomerulonephritis) and blood vessels (vasculitis) of various organs.
During targeted examination, Sjögren's syndrome is detected in 70-100% of patients with biliary cirrhosis. Involvement of the lacrimal and salivary glands in Sjögren's syndrome most often clinically manifests itself as keratoconjunctivitis sicca, xerostomia, decreased tear production during the Schirmer test, recurrent mumps and dry skin.
The pulmonary syndrome observed in patients with biliary cirrhosis is more radiological than clinical, and is characterized by a picture of diffuse pneumosclerosis with deformation of the pulmonary pattern due to additional stringy, looped and cellular tissues of the interstitial type and fibrosing alveolitis.
Accompanying illnesses
PBC is combined with other chronic diseases, mainly of an autoimmune nature: scleroderma, rheumatoid arthritis, Hashimoto's thyroiditis, myasthenia gravis, adult celiac disease, and transverse myelitis. Combined autoimmune disorders are naturally more common in women than in men. The incidence of insulin-dependent diabetes mellitus is higher in men than in women.
Scleroderma. The combination of PBC with scleroderma, according to various authors, ranges from 3 to 18%. In some cases, the clinical manifestations of scleroderma correspond to CREST syndrome (calcinosis, Raynaud's syndrome, esophageal dysfunction, sclerodactyly, telangiectasia). The pathological process involves the skin, mucous membranes, joints, and muscles. When scleroderma and PBC are combined, clinically significant lesions of internal organs are usually absent, which determines the benign course of the disease. Antinuclear antibodies and rheumatoid factor are usually determined in the blood.
Systemic lupus erythematosus. The variety and severity of manifestations are characteristic: skin, joint, muscle syndromes, lymphadenopathy, polyserositis, damage to the kidneys, lungs, heart, nervous system, hemocytopenia. Disease progression usually leads to death of patients 37 years after the onset of symptoms. LE cells and antibodies to native DNA are detected in the blood.
Rheumatoid arthritis. The incidence of rheumatoid arthritis in patients with PBC is up to 10%. The interphalangeal, wrist, knee, and ankle joints are mainly affected. The main symptoms are pain and swelling of the joints, impaired mobility in them, generalized lymphadenopathy, muscle atrophy in the area of the affected joints. X-ray examination reveals osteoporosis of the bones of the involved joints, narrowing of the interarticular spaces, and abnormalities of the articular surfaces. Rheumatoid factor is determined in serum, joint fluid, and also using an immunofluorescence reaction in the area of lymphoid infiltration of the synovial membrane.
Damage to the thyroid gland, according to various authors, with PBC is observed in 1832% of cases. The vast majority of patients have a clinical picture of hypothyroidism. We observed a combination of Hashimoto's thyroiditis with PBC in 3 women aged 4852 years. Significant enlargement and thickening of the thyroid gland, diffuse and nodular, appeared in 2 patients against the background of cirrhosis, and in one 1 year before the development of cholestasis. Antithyroglobulin and antimicrosomal antibodies are mainly determined in the blood.
Other autoimmune diseases can also be combined with PBC: autoimmune thrombocytopenia, fibrosing alveolitis, pernicious anemia, sarcoidosis, renal tubular acidosis. Of the skin lesions with a presumed immune pathogenesis, the most commonly associated with PBC is lichen planus.
The development of an immunodeficiency state, especially in cases of immunosuppressive therapy, is associated with a high incidence of malignant tumors of extrahepatic localization in patients with PBC. Breast cancer is detected in women with PBC 4.4 times more often than in the general population.
Laboratory data
Already in the early stages, an increase in the activity of cholestasis enzymes is characteristic:
ALP, leucine aminopeptidase, glutamyl transpeptidase. An increase in serum bilirubin level by 1.53.5 times compared to the norm is observed later and increases slowly. The concentration of bile acids and copper content in the blood serum increases, and the level of iron decreases. Already at the onset of the disease, pronounced hyperlipidemia is characteristic with an increase in the concentration of cholesterol, lipoproteins, phospholipids and non-esterified fatty acids. Serum aminotransferase values were increased 23 times, their activity correlated with histological data.
Particular importance in the diagnosis of PBC is given to AMA
. Currently, antibodies to 9 antigens of the inner and outer mitochondrial membrane are known. Of these, antiM2, M4, M8, M9 are associated with PBC. The remaining antibodies are associated with other diseases: antiM1 with syphilis, antiM5 with connective tissue diseases, antiM3 with drug-induced hepatitis, antiM7 with myocarditis. Antibodies to the mitochondrial inner membrane antigen M2 are found in almost all cases of PBC and are considered pathognomonic for this disease. AMA to M4 is detected in a disease with features of both PBC and autoimmune hepatitis (overlapsyndrome), to M8 in a rapidly progressive form of PBC, to M9 in the early stages of PBC.
Antimitochondrial antibody titers often correlate with PBC activity. AMA can be detected at the preclinical stage and does not disappear throughout the entire period of the disease.
Diagnosis
It is necessary to take into account gender, age, heredity, it should be especially emphasized that in 1/3 of cases the disease is diagnosed in women over 60 years of age
.
The most important clinical symptom is skin itching. In the early stages of the disease, the activity of cholestasis enzymes is increased, and an acceleration of ESR is noted. Antimitochondrial antibodies class M2 are a specific and valuable diagnostic test
. Ultrasound and CT scans reveal unchanged extrahepatic bile ducts.
The diagnosis is confirmed by histological examination of a liver biopsy, which reveals non-purulent destructive cholangitis in the early stages of the disease, and later the formation of biliary cirrhosis.
Diagnostic criteria for PBC:
1. Intense skin itching, clinical suspicion based on the presence of extrahepatic manifestations (sicca syndrome, rheumatoid arthritis, etc.).
2. Increase in the level of cholestasis enzymes by 23 times compared to the norm.
3. Normal extrahepatic bile ducts on ultrasound.
4. Detection of antimitochondrial antibodies in a titer above 1:40.
5. Increased level of IgM in blood serum.
6. Characteristic changes in the liver punctate.
The diagnosis of PBC is made in the presence of the 4th and 6th criteria or 34 specified signs.
Differential diagnosis
PBC must be distinguished from a number of diseases accompanied by hepatobiliary obstruction or cholestasis [2].
The most important diseases with which PBC is differentiated in adults:
- obstruction of extrahepatic bile ducts: stones, strictures, tumors;
- primary sclerosing cholangitis;
- carcinoma of the intrahepatic bile ducts;
- autoimmune hepatitis;
- drug-induced cholestasis;
- chronic viral hepatitis C;
- sarcoidosis
In childhood and adolescence, PBC is differentiated from:
- hypoplasia of the intrahepatic bile ducts,
- cholangiodysplasia (congenital liver fibrosis),
- biliary cirrhosis in cystic fibrosis.
It is most important to differentiate PBC from obstruction of the extrahepatic bile ducts, since patients with PBC often undergo unnecessary laparotomy for suspected subhepatic jaundice, and the correct diagnosis is made only after surgical liver biopsy
.
To differentiate PBC from obstruction of the extrahepatic bile ducts, primary sclerosing cholangitis, intrahepatic bile duct hypoplasia, congenital liver fibrosis, along with the study of antimitochondrial antibodies, direct visualization of the biliary tree
(endoscopic sonography, retrograde endoscopic or percutaneous transhepatic cholangiography).
Differential diagnosis in the early stages of PBC with autoimmune hepatitis in the absence of a clear histological picture in 15% of cases causes significant difficulties. However, the detection of such immunological phenomena as antimitochondrial antibodies of the M2 class, the predominance of IgM in the serum, and in liver biopsies the prevalence of damage to the bile ducts over changes in the liver parenchyma, destruction of the interlobular and septal ducts, makes it possible to diagnose PBC. Features of the disease such as high aminotransferase activity and detection of antibodies to smooth muscle can serve as guidelines for identifying autoimmune hepatitis.
In some cases, PBC must be differentiated from drug-induced chronic cholestasis. Unlike PBC, drug-induced cholestatic hepatitis occurs with less pronounced destruction of interlobular bile ducts and mild cellular infiltration of the portal tracts; antimitochondrial antibodies are absent; withdrawal of medications most often leads to a reversal of the process.
The greatest difficulties arise in distinguishing PBC from drug-induced cholestasis, accompanied by markers of autoimmunization. In liver biopsies in these cases, epithelioid cell and giant cell granulomas are often found, which differ from PBC in a large number of eosinophilic leukocytes. After drug withdrawal, the granulomatous reaction is replaced by fibrosis.
The prognosis depends on the stage of the disease. From the moment the first clinical signs of PBC appear, it is characterized by a gradual progression of the pathological process over 1220 years. Among the prognostic models, the most commonly used is the Mayo Clinic model, which takes into account age, bilirubin levels, serum albumin levels, prothrombin time and the presence of ascites [6]. The terminal stage is characterized by increasing liver failure, the appearance of ascites, hepatorenal syndrome, and encephalopathy.
Treatment
Advances in understanding the pathogenesis have led to attempts to use various drugs with immunosuppressive, anti-inflammatory, antifibrotic properties, as well as bile acids for the treatment of patients with PBC.
Glucocorticosteroids (GCS), prescribed at a dose of 30 mg/day for 8 weeks. with a gradual reduction of the dose to 10 mg/day, lead to an improvement in clinical symptoms, a temporary weakening of itching and/or increased fatigue, a decrease in the activity of aminotransferases, IgG, but do not affect the level of serum bilirubin. GCS cause a decrease in the inflammatory response according to liver histology. When placebo-controlled studies were continued for 2 years, no significant effect on mortality was noted. However, after a year of therapy, potentiation of osteoporosis became a big problem. Thus, GCS have potential value for the treatment of PBC, but the associated side effects make them considered dangerous substances and should not be prescribed for a long time in PBC. The risk of developing severe osteoporosis can be reduced by combining GCS with bisphosphonates.
Budesonide is a second-generation GCS with low systemic activity, causing virtually no side effects. The effectiveness of the drug in patients with PBC is being studied. There is reason to hope that this drug will be able to provide all the benefits of GCS without putting patients' lives at additional risk.
Cyclosporine A, a large European trial involving 349 patients with a follow-up of up to 6 years (mean 2.5 years), did not demonstrate prevention of histological disease progression or changes in survival of patients treated with the drug [5]. The high incidence of side effects, such as hypertension and deterioration of renal function, does not allow the drug to be used for the treatment of PBC.
Azathioprine, chlorambucil, malotilate, Dpenicillamine, due to the lack of a clear effect on the progression of the disease and the presence of serious complications, cannot be recommended for regular use in PBC.
Methotrexate at a dose of 15 mg orally once a week may have some effect on clinical symptoms, bilirubinemia and alkaline phosphatase activity. However, randomized controlled trials did not show an effect on the prognosis of the disease. Severe side effects were noted.
Colchicine is a prerequisite for the use of the drug due to its antifibrotic and anti-inflammatory effects. The drug's minimal toxicity has led physicians to recommend it for the treatment of PBC. In some cases, colchicine improves biochemical parameters. However, based on the results of randomized trials, it should be considered that colchicine does not have any effect on cholestasis, histological progression or patient survival.
The most promising drugs in the treatment of PBC include ursodeoxycholic acid and ademetionine.
Ursodeoxycholic acid (UDCA) is the drug that has undergone the most extensive efficacy studies in the treatment of patients with PBC. Of all pathogenetic therapy drugs, it is recognized as the most effective. Used at a dose of 1015 mg/kg for a duration of 10 months. up to 2 years or more, UDCA promotes the displacement of endogenous lipophobic toxic bile acids at the level of hepatocytes and biliary epithelium. This replacement of endogenous bile acids is due to competition between the polar hydrophilic ursodeoxycholic acid and these acids during their transepithelial transport in the ileum. A decrease in the amount of potentially toxic endogenous bile acids against the background of cholestasis is accompanied by a decrease in damage to cell membranes. In addition, UDCA is incorporated into the phospholipid layer of the cell membrane, which leads to a direct stabilizing effect on hepatocytes.
The immunomodulatory effect of UDCA is carried out by reducing the expression of MHC class I and II antigens on hepatocytes and epithelial cells of the bile ducts; UDCA reduces the synthesis of IL2, which leads to the suppression of stimulation of cytotoxic lymphocytes by type 1 helper cells.
Finally, the positive effect of UDCA is explained by its choleretic, hypocholesterolemic and litholytic effects.
UDCA contributes to a significant improvement in functional indicators, skin itching decreases or disappears. The effect on morphological parameters is ambiguous, since in some cases they can progress.
A combined analysis of French, American and Canadian trials was conducted, including 553 patients (276 receiving UDCA and 277 placebo). The average follow-up time was 4 years [4]. The results of the analysis showed that during UDCA therapy, liver transplantation was not required for a significantly longer period of time.
. In a multicenter trial in the United States, observed survival after two years of UDCA therapy was significantly higher than predicted [3].
Resistance to UDCA therapy requires the exclusion of other causes of liver damage, and first of all, the cross-talk between PBC and autoimmune hepatitis.
In all studies conducted, it was noted that beneficial effects were quickly achieved in the early stages of cirrhosis; UDCA can be considered as the drug of choice in the treatment of stage IIII PBC.
Ademetionine (Sadenosyl-methionine) is the initiator of three important metabolic pathways in the human body: remethylation, transsulfurization and polyamine synthesis. In these metabolic reactions, the drug acts either as a methyl group donor or as an enzyme inducer.
One of the most important factors in regulating the metabolic functions involved in the process of bile formation is the structure and composition of the hepatocyte membrane. With intrahepatic cholestasis, reduced membrane viscosity (a consequence of excess cholesterol deposition in it) leads to disruption of the functioning of protein transport systems localized in it. Ademetionine, participating in transmethylation reactions, one of which is the synthesis of phosphatidylcholines, increases membrane mobility and increases their polarization, which, in turn, leads to improved functioning of bile acid transport systems associated with hepatocyte membranes.
Liver transplantation is the treatment of choice for patients with progressive PBC and clinical and laboratory signs of hepatic decompensation. At the same time, the right moment for surgical intervention must be determined, since “major surgery” is unacceptable in patients with end-stage liver failure. Disabling weakness, refractory itching, and severe osteoporosis may be indications for inclusion on the waiting list at earlier stages of PBC. A successful transplant can restore full health for ten years or more, but sometimes PBC can occur in the transplanted liver.
Professor S.D.
Podymov MMA named after I.M.
Sechenov And also here!
Back
Causes:
Due to their occurrence, there are 2 types of biliary cirrhosis:
- Primary biliary cirrhosis of the liver
- Secondary biliary cirrhosis of the liver
Primary biliary cirrhosis - the mechanism of occurrence is that autoimmune inflammation occurs in the liver tissue itself. Antibodies are produced against liver cells (hepatocytes) and they are perceived by the human body as foreign. The process itself is joined by a protective system, in the form of lymphocytes, macrophages, mast cells, which produce biologically active substances and antibodies. All of them together destroy hepatocytes, cause disturbances in blood supply, metabolism and bile stagnation, leading to general destruction of the architectonics (structure) of the liver.
- Genetic predisposition
- People suffering from autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, thyrotoxicosis, scleroderma, polyarteritis nodosa, sarcoidosis
- It is known from scientific sources that about 15% of cases of the disease are caused by an infectious substrate such as the herpes virus, rubella, Epstein-Barr
Secondary biliary cirrhosis of the liver occurs due to blockage or narrowing of the lumen of the intrahepatic bile ducts.
- Developmental anomalies (congenital or acquired) of the bile ducts and gallbladder
- Cholelithiasis
- Narrowing or blockage of the biliary tract after surgery on the abdominal organs, benign tumors
- External compression of the biliary tract by an inflamed pancreas or tumor
Complications
Intrahepatic cholestasis in primary biliary cirrhosis can cause a lack of bile in the intestine, which can be accompanied by steatorrhea (an increase in the amount of fatty components in the stool), the development of a deficiency of fat-soluble vitamins, impaired bone mineralization and, as a consequence, the occurrence of osteoporosis. As liver disease progresses, patients gradually lose weight and usually develop symptoms such as blurred vision, bone pain, bleeding gums, and possible pathological fractures.
Symptoms of biliary cirrhosis of the liver
Main nonspecific symptoms:
- Weakness
- Headache
- Dizziness
- Decreased appetite
- Impaired memory and attention
- Weight loss
- Apathy
- Sleep disturbance at night and drowsiness during the day
Symptoms of hepatocellular failure:
- Vomiting of intestinal contents
- Intense pain in the right hypochondrium
- Bloating
- Alternating diarrhea and constipation
- Jaundice (yellowing of the skin and mucous membranes)
- Itchy skin
- Darkening of urine
- Stool discoloration
- The appearance under the skin of the eyelids, ears and phalanges of the fingers of yellow tuberous inclusions (xanthemas)
- Enlarged liver and spleen
Symptoms of portal hypertension:
- “Jellyfish head” is a symptom that combines the presence of an enlarged abdomen and the presence of a pronounced venous network on the skin of the anterior abdominal wall
- Vomiting “coffee grounds” is a symptom that indicates bleeding from the veins of the esophagus or stomach
- “Tar” stool is a symptom indicating bleeding from the small intestine
- Dark red blood released from the rectum during defecation is a symptom indicating the presence of bleeding from hemorrhoidal veins located in the rectum
- Palmar erythema - redness of the palms
- The appearance of telangiectasias on the skin - spider veins
Treatment
Pathogenetic therapy
. This treatment is prescribed to stop the progression of primary biliary cirrhosis, improve the patient’s quality of life and increase its duration. As part of pathogenetic therapy, ursodeoxycholic acid preparations, which have anticholestatic, cytoprotective, immunomodulatory and hypocholesterolemic effects, can be used. The drug Phosphogliv®1 can be used as part of complex therapy for cirrhosis.
Use of corticosteroids
. To achieve regression of the clinical symptoms of the disease, corticosteroid hormones (prednisolone or its analogues) can be prescribed. The use of such drugs can improve biochemical and histological parameters and increase life expectancy.
Symptomatic therapy
. To reduce skin itching, sorbents and agents are usually prescribed that can reduce the flow of bile acids into the enterohepatic circulation. For steatorrhea and bile deficiency in the intestines, enzyme preparations with high levels of lipase can be used. If the patient is experiencing severe bone pain, parenteral administration of calcium supplements may be indicated. They are also used to prevent and treat osteoporosis.
Surgery
. Liver transplantation may be prescribed if the level of serum bilirubin exceeds 150 µmol/l, there are signs of decompensation of the process (hepatic encephalopathy, repeated bleeding from the esophageal veins, etc.). Before the development of cirrhosis, surgical treatment can be used in connection with severe osteoporosis, intolerable skin itching, and a significant decrease in quality of life.
1 According to the instructions for medical use of the drug.
Reasons for development
It has not yet been possible to establish the specific cause of the formation of biliary cirrhosis. Some theories of its formation are considered:
- hereditary predisposition to the disease;
- previous infectious liver lesions - viral, bacterial, parasitic;
- toxic liver damage;
- autoimmune inflammation.
It is currently impossible to confirm a direct connection between these conditions and the formation of cirrhosis.
First, under the influence of certain reasons, lymphocytes begin to destroy the cells of the bile ducts - an inflammatory process is formed in them. Due to inflammation, the patency of the ducts is disrupted and bile stagnation develops. In these areas, hepatocytes are damaged and inflammation develops again. Massive cell death can lead to the formation of cirrhosis.
Indications for hospitalization of a patient with primary biliary cirrhosis
- first established diagnosis - clarification of the diagnosis and treatment (hospitalization of patients in the gastroenterological departments of the State Health Organization, Public Health Organization);
- the need to perform a liver biopsy (hospitalization of patients in the surgical departments of the Regional Healthcare Organization, State Health Organization, Public Health Unit);
- with the development of liver cirrhosis, indications for hospitalization are established by the protocol for this disease.
The basis for referring a patient for consultation to the Republican Scientific and Practical Center for Organ and Tissue Transplantation is the corresponding indications.
Diet
Diet plays an important role in the treatment of biliary cirrhosis. With the development of this disease, the functioning of not only the liver, but also other internal organs fails. And in order to reduce the burden on them and prevent the development of complications, all patients, without exception, need to follow certain rules in their diet. Depending on the severity of the disease and its accompanying complications, patients are assigned to treatment table No. 5a or No. 10 (mainly used when ascites occurs).
Violation of nutritional rules in biliary cirrhosis of the liver shortens the patient’s life
The daily calorie intake should not exceed 2900 kcal. In this case, it is imperative to refuse:
- fatty and fried foods;
- alcoholic drinks;
- dairy products;
- honey;
- fatty fish and meat;
- seafood;
- nuts
The main part of the diet should be fresh vegetables and fruits. You need to eat food in small quantities, but often - about 5-6 times a day. The last meal should be 2-3 hours before bedtime.
Food should be consumed boiled. Some dishes can be steamed. It is not advisable to bake food in the oven even without using oil and spices. In this case, food should be eaten warm. If liver function is impaired, hot and cold foods and drinks should not be consumed. Fasting days are held once every 2 weeks, during which the patient must eat only fresh vegetables and fruits.
Vegetables and fruits provide the body with vitamins and minerals, maintaining its functionality at the proper level.
An important point is compliance with the drinking regime. This allows you to avoid the occurrence of edema and complications in the form of disorders of the urinary system. Patients with biliary cirrhosis are recommended to drink at least 2 liters of purified water per day (this volume does not include teas and liquid dishes).
Classification of the disease
Biliary cirrhosis of the liver is assessed using the Child-Pugh scale, which implies an assessment of the functioning of the organ and:
- the presence of ascites and encephalopathy (one of the most common complications of liver diseases);
- bilirubin level;
- albumin concentration;
- prothrombin index, which indicates the presence of blood clotting disorders.
The difference between a healthy liver and a liver affected by cirrhosis
After studying all these indicators, points are given (from 0 to 10), then they are summed up and the class of biliary cirrhosis is determined. In total, there are 3 classes of this disease:
- A – it is characterized by minimal scores, the average life expectancy after surgery is 20 years;
- B – the number of points is average, after surgical interventions, life expectancy is on average 10 years, strip operations end in death in 30% of cases;
- C – high score, surgical interventions are rarely performed, abdominal operations end in death in 80% of cases.
There is also another classification of biliary cirrhosis of the liver - METAVIR, which is used to determine the degree of damage to the organ by fibrous tissue. This assessment is carried out through a biopsy. Gradation of degree - from 0 to 4.
Diagnostics
Diagnosis of biliary cirrhosis begins with a survey and examination of the patient. The doctor listens carefully to the patient for complaints, and then palpates the abdominal cavity, paying special attention to the area where the spleen is located. Then a biochemical blood test is prescribed.
With the development of biliary cirrhosis of the liver, there is an increased level of alkaline phosphatase and G-GTP, a violation of the ratio of ALT to AST, and an increase in the concentration of bilirubin. When examining the patient’s immune status, the following are observed:
- increased levels of rheumatoid factor;
- increased levels of antibodies to smooth muscle fibers;
- increased levels of antibodies to thyroid tissue;
- the presence of antinuclear antibodies in the biological material under study;
- the presence of antibodies to mitochondria in the blood;
- increase in the level of immunoglobulin M.
The results of laboratory blood tests are not the basis for making a diagnosis, but serve as a reason for further examination of the patient
If laboratory tests show abnormalities in the functioning of the immune system and liver functionality, a computer study is performed to confirm the diagnosis - ultrasound and CT of the hepatobiliary system. These diagnostic methods make it possible to verify the development of liver fibrosis. However, an accurate diagnosis can only be made based on the results of a biopsy.
But there is one nuance here too. A diagnosis of biliary cirrhosis of the liver can only be made during a biopsy in the initial stages of the disease. Further, the morphological signs of the disease become similar to all types of cirrhosis.
Since the development of biliary cirrhosis, in addition to autoimmune processes, can provoke other diseases in the body, to obtain a more complete picture, ultrasound of all internal organs of the abdominal cavity, ERCP and MR cholangiopancreatography are also performed.