Sickle cell anemia refers to a group of genetic disorders that affect hemoglobin. The disease can be life-threatening, but there are ways to manage the symptoms. Red blood cells contain hemoglobin, which delivers oxygen to the body's tissues. In sickle cell disease, the red blood cells are C-shaped (like a sickle) and sticky. As a result, red blood cells can block blood vessels and are unable to effectively deliver oxygen, resulting in a variety of symptoms.
Genetics Home Handbook , sickle cell disease disproportionately affects “people whose ancestors come from Africa; Mediterranean countries such as Greece, Türkiye and Italy; Arabian Peninsula; India and the Spanish-speaking regions of South America, Central America and parts of the Caribbean."
Types
There are several different types of sickle cell disease. The main types include:
- HbSS
- Sickle cell disease (homozygosity for abnormal HbS): A person inherits two genes, one from each parent, for sickle cell disease. He will have sickle cell anemia, which is the most severe type of sickle cell anemia. - HbSC is a double heterozygous (compound heterozygous) carrier of two abnormal hemoglobins HbS and HbC
: a person inherits the sickle cell gene from one of the parents. A gene is inherited from the other parent that results in a different type of abnormal hemoglobin. This disease is usually less serious. - Sickle cell beta thalassemia HbS
: A person inherits the sickle cell gene from one parent and the gene for beta thalassemia, another type of anemia, from the other parent. - Carriage of the sickle cell trait
: If a person has only one gene, they will not have sickle cell disease, but they can pass the gene on to their children.
Sickle cell disease can affect anyone, but is more common in black people. Sickle cell anemia is more likely in areas where malaria is common. However, patients with sickle cell disease have a lower risk of some severe types of malaria.
Causes
The causes of this serious illness lie in the physicochemical properties (solubility, erythrophoretic mobility) of hemoglobin, but not that which is recognized as normal (HbA), capable of providing respiration and nutrition to tissues. It's all about the abnormal hemoglobin HbS, which was formed as a result of a mutation and is present in patients with this disease instead of the normal one - HbA. By the way, hemoglobin S became the first described and deciphered red blood pigment of all defective hemoglobins (there are other abnormal Hb, for example, C, G San Jose, which, however, do not give such a severe pathology).
abnormal structure of the amino acid chain of hemoglobin S
Since the disease is inherited from parents to children, it was natural to assume that the cause of such a defect is some gene that arose during a pathological mutation. (gene mutations occur constantly - both beneficial and harmful). Research at the genetic level has shown that this is indeed the case. The anomaly has found its place in the structural gene of the beta chain; because of it, one base is replaced in the 6th amino acid by another (adenine to thymine). However, to make it easier for the reader to understand, there is a need to very briefly explain some of the terms used by geneticists, otherwise the reasons for the appearance of such a severe pathology will remain unclear. Thus, the sequence of amino acids in a protein (and hemoglobin, as we know, is a protein) encodes 3 nucleotides, which are controlled by certain genes and are called coding trinucleotides, codons or triplets.
In this case it turns out that:
- The gene, untouched by the mutation (triplet in healthy people - GAG), ensures the formation of normal hemoglobin (HbA);
- In patients, as a result of a point mutation, the appearance of a pathological gene for sickle cell anemia, adenine is replaced by thymine and the encoding trinucleotide is already GTH.
Due to such changes at the genetic level, such bad consequences arise as:
- Replacement of negatively charged glutamic acid with neutral valine;
- A change in the charge of the HbS molecule, and, therefore, in the physicochemical properties of hemoglobin.
inheritance of sickle cell anemia (autosomal recessive type with incomplete dominance)
Sickle cell anemia - symptoms
If the body's cells do not receive enough oxygen, various symptoms and complications develop. It can happen at any age and symptoms will vary from person to person. Sickle cells are destroyed more easily than healthy red blood cells. This can lead to low levels of red blood cells (RBCs), known as anemia.
Early symptoms may include:
- jaundice, or yellowing of the skin and whites of the eyes
- fatigue
- pain and swelling in the arms and legs
Further symptoms and complications may include:
- episodes of pain
- swelling in the arms and legs
- acute chest syndrome
- vision loss
- enlarged spleen
- leg ulcers
- stroke
- deep vein thrombosis
- damage to the liver, heart or kidneys
- cholelithiasis (in young people)
- infertility (in men)
- pulmonary hypertension, which is high blood pressure in the blood vessels supplying the lungs
- heart failure
- damage to bones and joints that occurs due to low blood supply
- high risk of infections, which may have severe symptoms
- fever
The main symptoms and complications of sickle cell anemia:
- Pain
: During a painful episode, the sickle cells become clogged and prevent blood flow to any part of the body. The pain can range from mild to severe and can last for any period of time. - Infections
: People with sickle cell disease may be at high risk of severe illness from COVID-19, regardless of age. - Chest syndrome
- includes chest pain, cough, fever and difficulty breathing. - Enlarged spleen
: weakness, pale lips, rapid breathing and heart rate, thirst and abdominal pain are noted.
If any of the above symptoms appear, a person needs emergency medical attention.
In infants
Because a person inherits the disease, symptoms of sickle cell disease are observed before birth. A routine blood test at birth will show whether this condition is present. In severe cases, the child may develop aplastic crisis, in which the bone marrow stops producing red blood cells, resulting in severe anemia. The child may have an enlarged spleen. Symptoms include poor appetite and lethargy.
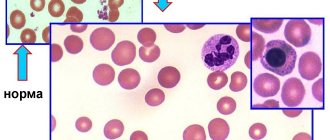
Clinical blood test
Suspecting absolutely any blood disease, doctors refer patients for a clinical (general) blood test.
It helps determine whether organs are in normal condition, and also provides information about proper hematopoiesis and the presence of infectious infection. For this type of analysis, both blood from a finger and from a vein are suitable.
When drawing blood from a finger with suspected sickle cell anemia, it is wound with a tourniquet, creating conditions of oxygen deficiency. This provokes a large influx of red sickle-shaped cells to the finger, which helps make the diagnosis easier.
Abnormalities that occur in the blood during SCD are shown in the table below (Table 1):
Table 1
| Component name | What does this mean? | Generally accepted standards | Deviations in SCA (HA – hemolytic crises) |
| (RBC) Red blood cells | A reduced level of red blood cells in the body is associated with sickle cell anemia and increased death of red blood cells. During periods of GC, the condition is especially characteristic, since more than 50% of red cells can be neutralized. | Men (M): 4.0 – 5.0 x 10 12 /l. | Less than 4.0 x 10 12/l. |
| Women (F): 3.5 – 4.7 x 10 12 /l. | Less than 3.5 x 10 12 /l | ||
| (HGB) Hemoglobin | The total hemoglobin level drops due to the destruction of red blood cells. | M: 130 – 170 g/l. | Less than 130 g/l. |
| F: 120 – 150 g/l. | Less than 120 g/l. | ||
| (WBC) White blood cells | This indicator increases when the body is affected by infectious diseases, which is especially typical in sickle-shaped anemia. | 4.0 – 9.0 x 10 9 /l. | A white blood cell count above 9.0 indicates the presence of infection. |
| (PLT) Platelets | With a large size of the spleen, not only sickle-shaped, but also normal red blood cells are destroyed, this happens in the case of the development of hypersplenism in the blood. | 180 – 320 x 10 9 /l. | Within normal limits, or reduced. |
| (RET) Reticulocytes | A decrease in the quantitative indicator of red blood cells contributes to the production of erythropoietin by the kidneys, as a result of which the processes of blood formation in the bone marrow are enhanced. As a result, a larger number of reticulocytes are released into the peripheral bloodstream. | M: 0.24 – 1.7%. F: 0.12 – 2.05%. | Increased, especially BG. |
| (Hct) Hematocrit | The ratio of plasma elements and formed elements of blood shows hematocrit. Hemolysis entails a decrease in the total number of red blood cells, as a result of which the hematocrit index also drops. | M: 42 – 50%. F: 38 – 47%. | The deviation is below the norm by up to 20%, at the moments of the GK. |
| (ESR) Erythrocyte sedimentation rate | Red cells settle to the bottom of the tube at a certain rate. The low content of red blood cells in the body causes them to settle to the bottom of the test tube faster. | M: 3 – 10 mm/hour. F: 5 – 15 mm/hour. | More than 10 mm/hour. More than 15 mm/hour. |
Blood biochemistry
This type of blood test helps to more broadly determine the state of the body, gives indicators for almost all organs of the body, and for the quantitative and qualitative state of the blood.
In biochemistry, there are also normal indicators, deviation from which indicates a particular pathology.
The biochemistry study for SCD is given below (Table 2):
Table 2
| Name | What does this mean? | Normal indicator | Deviations in sickle cell anemia (HA - hemolytic crisis) |
| Bilirubin indicator | It shows how quickly the red ones in the spleen are destroyed. | 4.5 – 17.1 µmol/l. | Many times higher than normal. |
| Free plasma hemoglobin | SGP shows the rate of destruction of red blood cells in the vessels; during this process, multiple amounts of hemoglobin are released into the blood. | Less than 220 mg/l. | Many times higher than the norm, especially during the period of hysterectomy. |
| Plasma haptoglobin concentration | The protein formed in the liver is called haptoglobin. It is responsible for moving hemoglobin to the spleen for further destruction. When red blood cells die in the vessels, its hemoglobin levels increase, so haptoglobin falls. | 0.8 – 2.7 g/l. | Many times higher than the norm, especially during the period of hysterectomy. |
| Free iron level in blood | In the case of GC plaza, the quantitative indicator of free iron increases. The protein does not have time to remove all the iron that appears during the breakdown of multiple red blood cells, which increases the concentration of iron in the blood. | M: 17.9 – 22.5 µmol/l. F: 14.3 – 17.9 µmol/l. | At times of high blood pressure, this indicator can be several times higher than normal. |
| Alanine aminotransferase (ALAT) and aspartate aminotransferase (AST) levels | ALT and AST are enzymes stored in the liver in predominant quantities. The higher their indicator, the more liver tissue is destroyed. | M: up to 41 U/l. F: up to 31 U/l | In case of extensive liver damage, it is much higher than normal. |
Sickle cell anemia - diagnosis
Newborn screening involves a simple blood test to detect sickle cell disease and carrier status of the sickle cell trait. If tests show that your child has sickle cell disease, doctors will monitor him over time. Tests are also available before birth, starting at 8-10 weeks of pregnancy.
If a person with a family history of sickle cell disease plans to have children, they can see a genetic counselor and undergo genetic testing to find out if their children are likely to have the disease.
Sickle anemia: diagnosis and treatment
Only a hematologist can diagnose and treat sickle cell pathology. The diagnosis is not made only on the basis of external symptoms; it is necessary to collect a detailed family history, clarify the time and circumstances under which the signs of pathology appeared for the first time. But the diagnosis can only be confirmed through specific examinations:
Sickle cell anemia in one population is determined by:
- Traditional blood test.
- Blood biochemistry.
- Results of ultrasound, radiography.
There are no effective treatments that make it possible to completely get rid of this disease. The patient can only be helped by preventing an increase in the number of modified red blood cells. In addition, it is necessary to stop the external signs of the disease in a timely manner.
The principal treatment for this anemia consists of:
- Healthy lifestyle.
- Medicines that increase hemoglobin protein levels and increase the number of undeformed red blood cells.
- Oxygen therapy.
- Relief of local pain.
- Eliminate iron deficiency.
- Prevention of viral invasions.
A method that allows you to determine the percentage probability of inheritance of a pathology is PCR. The parental genetic material is examined and mutated regions of the genome are identified. The result is considered to be both their presence/absence and the determination of the type and form of the disease if present – homozygous/heterozygous anemia.
Sickle cell anemia - treatment
Drug treatment
The following medications may reduce the risk of complications:
- Hydroxyurea
(Hydrea): Helps provide oxygen to the body. It is not safe to use during pregnancy. Studies have not proven the benefit of this drug in children younger than 9 months. - L-Glutamine Oral Powder
(Endary): Helps reduce the number of sickle cells and is suitable from age 5. - Voxelomotor
(Oxbrita): Increases healthy hemoglobin levels. Suitable from 12 years of age. - Crizanlizumab-tmca
: Reduces pain by preventing blood cells from attaching to blood vessels. Suitable from 16 years of age.
Prevention of infections
Doctors recommend that adults and children take precautions to reduce the risk of infection. These include:
- regular hand washing
- stay away from reptiles such as turtles as they can carry salmonella
- Get vaccinations against influenza, pneumococcal disease, and meningococcal disease
Your doctor may prescribe penicillin or another antibiotic for children under 5 years of age.
Blood transfusion and stem cell transplantation
People with severe symptoms may require a blood transfusion or stem cell transplant. Blood transfusions may be necessary if the patient has severe anemia, an enlarged spleen, infection, or other complications.
A stem cell transplant using cells from a healthy donor can cure sickle cell disease, but can be risky. In addition, the stem cells must be precisely selected.
What are red blood cells and hemoglobin?
Globin chains are formed at the genetic level, and under the control of genes of different chromosomes.
Red blood cells called erythrocytes are responsible for moving oxygen cells around the body and delivering them to organs.
They are disc-shaped and small in size (7.5-8.3 microns), the red blood cell has increased thickness at the edges and a hollow in the center.
Due to this shape, it can pass through even the thinnest capillaries, the passage size of which is two to three times smaller than the diameter of the red cell.
The space inside the red blood cell is almost completely filled with hemoglobin. Which in turn consists of globin (a protein) and heme (an element containing iron).
Each individual red blood cell contains about 30 pg of hemoglobin.
As a result of the combination of amino acids (protein components), the alpha and beta chains of the globin protein are formed, two of each (a1 and a2 + b1 and b2).
In addition to the formation of alpha and beta protein chains, other globin chains (sigma, gamma and delta types) can also form in red cells.
The combination of these types in chains leads to the formation of different types of hemoglobin, inherent in certain stages of human development.
Other types of protein chains may be:
- HbA. The form of normal hemoglobin, more than 90% in adults, consists of two alpha and two beta protein chains;
- HbA A minority of hemoglobins, accounting for up to 2% of the total amount of human hemoglobin. Contains two alpha and two sigma protein chains;
- HbF. Also called fetal hemoglobin, it contains two alpha and two gamma chains and is primarily found during fetal development inside the womb. The levels of this protein in the blood of an adult do not rise above 1.5%;
- HbU. Called fetal hemoglobin, it is formed inside red cells after 2 weeks from the moment of conception and is completely replaced by HbF after the start of blood formation in the liver.
Lifestyle
Many people with sickle cell disease can live full and active lives, especially if they take steps to reduce the effects of the condition. Lifestyle measures that may help include:
- regular medical examinations
- drink 8 to 10 glasses of water per day
- do not overcool or overheat, as this can provoke a crisis
- engage in physical activity, but also get enough rest
- maintaining a healthy diet.
Scientific article on the topic: Gene therapy will help cure sickle cell anemia.