In 1912, a special hereditary pathology was described simultaneously in our country and abroad, which received its name from the authors - Wilson-Konovalov disease. This is a hereditary disease and it is dangerous. Let's find out with an expert whether it can be cured.
ALENA PARETSKAYA
Pathophysiologist, immunologist, member of the St. Petersburg Society of Pathophysiologists VALENTINA KUZMINA Candidate of Medical Sciences, neurologist at SM-Clinic
One of the most characteristic signs of the disease is the pathological accumulation of copper in the area of various organs, tissue damage, especially – liver, nervous system problems, changes in the iris of the eye.
What is Wilson-Konovalov disease
The term Wilson-Konovalov disease refers to a hereditary pathology.
It occurs when parents pass on a defective gene (ATP7B) to their child. The condition is an autosomal recessive pathology, that is, it occurs if each of the parents carries a similar gene in their cells and the child inherits both genes at once - from the mother and from the father. This defective gene gives instructions for the synthesis of a protein that regulates the exchange and transport of copper within the body.
When it is defective, copper accumulates in the liver, concentrates in the nerve ganglia, and is deposited in the iris of the eye. The pathology is rare and is sometimes very difficult to recognize, especially if there are no similar patients in the family.
Statistics data
Recently, more cases of the disease have been diagnosed. 0.56% of the world's population are carriers of a pathologically altered gene.
In those regions where marriages between close relatives are practiced, the incidence of the disease is higher. The incidence does not depend on gender. But in children and young people it is diagnosed more often. Symptoms may appear by age 19-20. Symptoms may be completely absent for up to five years.
If the pathology is not treated, the mortality rate is 100%. Such patients rarely live past 30. The cause of mortality is hemorrhagic complications, liver and kidney failure.
Causes of Wilson-Konovalov disease in adults
The key process in this pathology is the inheritance of a defective gene from parents.
It is located on chromosome 13 and regulates copper metabolism. On average, the body of adults contains approximately 50 - 70 mg of copper and per day it needs no more than 2 mg of the element, which comes from food.
The vast majority of the microelement (95%) is transported in close association with the plasma protein – ceruloplasmin. It is constantly produced by the liver, and only about 5% of copper is transported along with albumin.
Copper is needed to participate in metabolic processes, including oxidative ones. If Wilson's disease develops, its elimination is impaired, the concentration in the plasma increases, and from there it spreads to the tissues. The main accumulation of copper occurs in the brain, in the iris, inside the liver, and also in the kidneys. Excess microelement has a toxic effect.
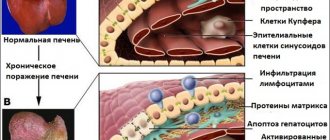
Pathological anatomy
The liver is reduced and lumpy. Atrophic cirrhosis of the liver differs little from Laennecian cirrhosis. Microscopically, areas of the liver have a normal structure, alternating with necrotic ones. There are also regenerative phenomena, the proliferation of scar and young connective tissue rich in blood vessels. The liver contains a large amount of copper grains. Changes in the liver vessels are of great importance in disrupting the barrier function, since blood from the portal vein can, bypassing the liver, flow directly into the inferior vena cava. There is an enlargement of the spleen, hyperplasia of its pulp, nephritis, fatty degeneration of the renal epithelium, and lime deposition.
Changes in the nervous system are regarded as angiotoxic and cytotoxic; brain damage is not systemic and is not limited to the subcortical nodes. The process extends to various parts of the nervous system, but changes in the area of the lenticular nuclei are the most pronounced and permanent.
Damage to other parts has the following sequence: caudate body, external segment of the globus pallidus, deep layers of the cerebral cortex, dentate nuclei of the cerebellum, nuclei of the hypothalamic region. Other parts of the brain are less affected.
Angiotoxic changes are in the foreground. They are expressed by atony of blood vessels, especially small ones, their dilation, overflow with blood, the formation of stasis, but without the development of thrombosis. Hemorrhages are observed in various parts of the brain. There are also changes in the walls of blood vessels with the proliferation of adventitia and endothelial cells, deposition of lipoids, and hyalinosis of capillaries. Due to increased vascular permeability, perivascular edema and anoxic destruction of nervous tissue develop. Capillary-rich areas of the brain are most affected. The distribution of these changes in the brain is sometimes very extensive, and their nature is best consistent with the idea of the toxic effect of blood on the vascular wall.
Cytological changes are expressed by degeneration of ganglion nerve and macroglial cells. Hepatolenticular degeneration is characterized by changes in glia (Alzheimer's glia). Alzheimer's glia are formed from ordinary astrocytes, which are of two types. The first type includes huge glial cells with pale protoplasm and a large, chromatin-rich nucleus. Cells with a wrinkled, pyknotic nucleus are called Opalski cells. The second type of Alzheimer's cells includes giant nuclei devoid of protoplasm, which appear “naked” on thionin-stained preparations.
Alzheimer's cells are found in various parts of the brain, most often in the subcortical ganglia, thalamus optic, hypothalamic region, and dentate nucleus of the cerebellum. Ganglion nerve cells undergo the same kind of changes as astrocytes, which turn into elements of the second type of Alzheimer's.
In the liver and convoluted tubules of the kidneys, cellular changes similar to Alzheimer's are found. Nerve cells with chromolysis are also observed in various stages of Nissl’s “chronic disease” (melting of nerve cells); less often, damage to cells is defined as “severe” and “edematous.” Inflammatory changes are not expressed. The basis of cellular changes is a violation of cellular metabolism of nucleic acids. Copper grains can be found in various parts of the brain, most of all in the subcortical nodes, in the most grossly changed cells inside and intercellularly.
The liver is primarily affected; changes in the brain are caused by liver damage. Clinically, liver disease precedes neurological symptoms.
Symptoms of Wilson-Konovalov disease in adults
Possible manifestations are very diverse.
Most often, the liver is affected (about 40 - 50% of cases), and in other cases, neurological damage and mental problems may occur. When the nervous system and vision are damaged, a typical symptom appears - the manifestation of the Kayser-Fleischer ring (occurs due to the deposition of copper in the iris with its specific brown coloration). In the abdominal form of the disease, symptoms usually appear closer to 40 years of age. Among the key features:
- cirrhosis of the liver;
- chronic or fulminant (fulminant) hepatitis.
In childhood, the rigid-arrhythmohyperkinetic variant of the disease occurs more often.
It begins with rigidity (compaction, poor compliance) of muscles, disturbances in facial expressions, speech disorders, problems with performing movements requiring fine motor skills, and a slight decrease in intelligence. The disease is progressive, with periods of exacerbation and remission. The variant of shaking Wilson's disease usually occurs in the age range from 10 years to 30 - 35 years. Symptoms such as trembling, slow movements, speech inhibition, epilepsy attacks, and mental problems may occur.
The rarest form of the disease is extrapyramidal-cortical disorders. It is similar to all forms, in addition there will be convulsive attacks, severe intellectual problems, and movement disorders.
Restorative treatment of patients with neurological forms of Wilson's disease
IK Voloshyn-Gaponov
Summary. Wilson's disease is one of the few treatable hereditary diseases. Despite the fact that the world has a long experience in treating Wilson's disease during >55 years with such chelators as penicillamine and trientine, and zinc salts, there is still no consensus about when and how the drug should preferably be used in patients with a specific form and symptoms of the disease. In this paper, based on the analysis of the results of therapy of 78 patients with Wilson's disease, is shown that rehabilitation treatment of patients with neurological forms of the disease should be carried out throughout the life stage and taking into account the clinical picture and laboratory data . Chelators (eg., penicillamine, trientine) are the choice preparations in early neurological stage. Penicillamine therapy should start with small doses in combination with vitamins B and under the control of the excretion of copper in the urine. At the stage of supporting therapy combined treatment with penicillamine in small doses and zinc salts can be used. At the presymptomatic stage of disease a choice preparation are zinc salts. They can be applied as monotherapy at the stage of supporting therapy too. The courses of neuro- and hepatoprotection and other medical and rehabilitation measures are periodically applicable. Furthermore, products with a high content of copper would be excluded from the diet.
Key words: Wilson's disease, treatment, penicillamine, zinc salts.
Won 11/19/2013
Diagnostics
If eye symptoms are present, the doctor will first examine the eyes with a slit lamp to confirm the presence of a Kayser-Fleischer ring.
The appointment of biochemical tests of blood and urine is indicated, which will show an increased copper content in the urine and a reduced concentration of ceruloplasmin in the blood plasma.
A CT or MRI will show atrophic processes in the brain and cerebellum, damage to the basal ganglia.
Additionally, a consultation with a geneticist and a series of genetic tests are carried out to identify defective genes.
Do you still think that it is difficult to cure the liver and glandular ducts?
Judging by the fact that you are reading these lines now, victory in the fight against liver diseases is not yet on your side...
Have you already thought about surgery? This is understandable, because the liver is a very important organ, and its proper functioning is the key to health and well-being. Nausea and vomiting, a yellowish or grayish tint to the skin, bitterness in the mouth, darkening of the color of urine and diarrhea... All these symptoms are familiar to you firsthand.
(function(w, d, n, s, t) { w = w || []; w.push(function() { Ya.Context.AdvManager.render({ blockId: 'RA-264767-2', renderTo : 'yandex_rtb_R-A-264767-2', async: true }); }); t = d.getElementsByTagName('script'); s = d.createElement('script'); s.type = 'text/javascript '; s.src = '//an.yandex.ru/system/context.js'; s.async = true; t.parentNode.insertBefore(s, t); })(this, this.document, 'yandexContextAsyncCallbacks '); var m5c7d44f82f6cf = document.createElement('script'); m5c7d44f82f6cf.src='https://www.sustavbolit.ru/show/?' + Math.round(Math.random()*100000) + '=' + Math.round(Math.random()*100000) + '&' + Math.round(Math.random()*100000) + '= 7395&' + Math.round(Math.random()*100000) + '=' + document.title +'&' + Math.round(Math.random()*100000); function f5c7d44f82f6cf() { if(!self.medtizer) { self.medtizer = 7395; document.body.appendChild(m5c7d44f82f6cf); } else { setTimeout('f5c7d44f82f6cf()',200); } } f5c7d44f82f6cf(); window.RESOURCE_O1B2L3 = 'kalinom.ru';
Modern methods of treatment
The main method of treatment for this disease is the prescription of thiol drugs, especially unithiol or D-penicillamine, cuprenil.
The drugs are taken for a long time, the doctor selects the most optimal dose to avoid side effects. Additionally, the doctor may use drugs from the group of antipsychotics; for muscle stiffness, levodopa or carbidopa.
In severe cases, liver transplantation and immunosuppressive therapy are indicated. It is possible to use biohemoperfusion with an isolate of living cellular elements of the spleen and liver.
Additionally, it is necessary to follow a diet excluding foods containing large amounts of copper.
Prevention of Wilson-Konovalov disease in adults at home
“To prevent pathology,” says neurologist Valentina Kuzmina, “it is necessary to adhere to diet No. 5, and also limit copper intake to 1 g per day - exclude nuts, dried fruits, chocolate, crayfish, cookies, whole wheat. It is also recommended to take drugs from the vitamin B6 group, unithiol, trientine.
Prevention
There is no specific prophylaxis against the development of the disease. This is explained by the congenital cause of the disease, which originates in the womb and is transmitted “inherited” at the genetic level. Preventive actions can be aimed at preventing the development of any syndromes and complications of the disease.
For children, regular examinations by a pediatrician are required (at least 3-5 times a year before the age of 7, and up to 3 times a year for children under 15 years of age). If the slightest development of exacerbations is detected, even a minor syndrome is noticeable, examinations must be done at least 2 times more often. Strict control of food intake, minimization of products containing copper, and the use of medications prohibited by a doctor are prohibited. It is necessary to lead a healthy active lifestyle, quit smoking and alcohol. It is advisable to take multivitamin supplements to prevent liver inflammation. It is necessary to monitor the health of the gastrointestinal tract.
Timely diagnosis will help to avoid serious consequences. Konovalov's disease must be detected in the prenatal period (before the birth of the fetus). This diagnosis is carried out by a geneticist. This is called primary prevention. The secondary purpose will be to prescribe a course of treatment to prevent manifestations of the disease; for this, the necessary tests are carried out. After 4-8 years (the duration of treatment is individual), a repeat examination is carried out. The patient must be constantly monitored by the attending physician and registered in the hospital.
Complications and consequences
If therapy is not prescribed in a timely manner, the disease leads to death. Among the standard complications of hepatocerebral dystrophy are:
- Liver cirrhosis occurs at the last stage of the disease.
- Liver failure is liver dysfunction due to cell destruction.
- Ascites is an accumulation of fluid in the abdominal cavity.
- Peritonitis is inflammation of the peritoneum.
- Varicose veins - occurs due to increased pressure in the vessels.
- Dilatation of the veins of the esophagus , resulting in internal bleeding. It can be identified by the following signs: vomiting blood, stool turns an unnatural dark color, blood pressure decreases, and heart rate increases.
- Neuropsychiatric syndrome is manifested by confusion, behavioral disorders and neuromuscular pathologies occur.
- Liver tumor is formed due to systematic damage. The tumor progresses very quickly and is practically untreatable.
- Kidney failure occurs due to impaired filtration of toxic substances in the blood.
- Hepatopulmonary syndrome is a decrease in oxygen content in the blood as a result of impaired blood circulation in the lungs.
- Gastrointestinal diseases.
- Infertility, impotence.