Traumatologist-orthopedist
Shelepov
Alexander Sergeevich
12 years of experience
Doctor
Make an appointment
A less common genetic pathology characterized by changes in connective tissue is Marfan syndrome. People with classic signs of this disease are easy to recognize by their characteristic appearance. Almost all of them are characterized by abnormally high growth and asthenic physique, elongated limbs and fingers, and excessively mobile joints. The disease is characterized by various pathological changes in the structure of the skeleton, heart muscle and blood vessels, and organs of vision. The incidence of Marfan syndrome is low and amounts to one per 10-20 thousand newborns, and this indicator is not influenced by gender or race.
Features and causes of the disease
The initial signs of Marfan syndrome appear in the prenatal period of development. They are caused by disorders of the development of connective tissues, which are caused by a mutation of the gene that regulates the production of one of the main proteins - fibrillin. Due to structural changes and fibrillin deficiency, tissues become less dense and elastic and do not tolerate loads well. The joints and ligaments, the walls of blood vessels and the eye apparatus, in which the tissue of the ligament of zinn is weakened, suffer most severely because of this.
The main cause of Marfan syndrome is the autosomal dominant inheritance of the mutation, i.e. the disease is transmitted from one parent to the child. In addition, in some cases, changes in the gene structure appear due to the impact on a woman of external unfavorable factors, which include radiation, ionized radiation, as well as radiation therapy to which the mother was exposed during the treatment of cancer.
Doctors distinguish erased and pronounced forms of the disease. In an erased form, changes are present in one or two systems, and they are quite minor. In a severe form of the disease, changes are present in at least three systems, regardless of the degree of their severity, or in one or two, but are quite pronounced. The patient's condition may remain stable for many years, or the pathology may progress, covering new areas of the body, systems and organs.
Medical history
In 1876, symptoms of an unknown pathology were noted by Dr. Williams, but clinical observations were carried out much later - in 1896 by a pediatrician from France A. Marfan. For 5 years, the doctor assessed the condition of a girl with previously unstudied anomalies, consisting in the progression of skeletal and muscle tissue dystrophy.
By the mid-20th century, there were many described cases in which patients exhibited symptoms close to Marfan's pathology, and all of them were hereditary diseases. Among such cases are aortic dissection, heart defects, ectopia of the lens, accompanied by bone deformation (chest, spine) and external deviations from the norm (tall stature, thinness, long limbs). American geneticist McKusick conducted a detailed study of chromosome mutations and discovered a new group of connective tissue diseases.
Main signs of pathology
Often, external symptoms of Marfan syndrome appear already in the first days after the birth of a child and then only intensify. Among the external signs by which pathology can be suspected, it should be noted, first of all:
- increased length of limbs and fingers (dolichostenomelia and arachnodactyly);
- insufficient weight with increased physical development of the child;
- elongated skull shape and elongated face;
- weak, poorly developed muscle tissue, lack of fatty tissue;
- abnormally high joint flexibility;
- awkwardness and clumsiness of movements.
Marfan syndrome in children over four years of age leads to changes in the shape of the chest, curvature of the spine, and the development of flat feet.
Among the ophthalmological symptoms, the most common are myopia, ectopic lens, changes in the shape of the cornea, strabismus, hypoplasia of the iris and retina. Changes often appear already in the first years of life and are bilateral in nature, steadily progressing over time.
The most dangerous are pathological changes in the cardiovascular system, which, in the absence of medical care, lead the patient to death at an early age. This includes changes in the vascular walls, various defects in the structure of the heart and coronary vessels. In the most unfavorable form of the disease, the child develops progressive heart failure already in the first year of life.
In addition, symptoms of Marfan syndrome may manifest themselves in the functioning of other systems and organs. The disease can affect nervous tissues, bronchi and lungs, skin, urinary and reproductive systems.
Are you experiencing symptoms of Marfan syndrome?
Only a doctor can accurately diagnose the disease. Don't delay your consultation - call
How to recognize Marfan syndrome
People with this disease are distinguished by their appearance as Marfan syndrome / Mayo Clinic. They usually have a tall and slender figure, disproportionately long arms, legs and fingers, a long face and very crowded teeth that can overlap each other. Often the sternum protrudes too much or, conversely, is pressed inward, and the spine is strongly curved.
In addition, there are other symptoms of Marfan Syndrome / National Institute of Arthritis and Musculoskeletal and Skin Diseases:
- Headache.
- Flat feet.
- Heart rhythm disturbances. It may beat more often or less frequently than normal, and sometimes misses beats.
- High palate.
- Loose, very mobile joints.
- Lower back pain, numbness in legs.
- Shortness of breath due to changes in the lungs and heart.
- Stretch marks on the skin.
- Poor eyesight.
How to accurately determine pathology in a child?
Currently, the diagnosis of Marfan syndrome is based on compliance of the clinical picture with the Ghent criteria, developed in 1995 and refined in 2010. They describe a number of signs of pathology for the skeletal system, organs of vision, heart and blood vessels, as well as for other systems and organs. To determine the degree of compliance, the doctor collects anamnesis (including family history), conducts a thorough examination of the patient with phenotypic tests, prescribes laboratory tests and instrumental studies, which include:
- urine test to determine glycosaminoglycans;
- identification of DNA genotype;
- conducting ECG and EchoECG to identify pathologies of the heart and blood vessels;
- performing ultrasound of the heart;
- chest X-ray to document deformities of the skeleton, heart and lungs;
- computed and magnetic resonance imaging.
If necessary, other tests and studies may be prescribed.
Pathogenesis
More than half of human weight is represented by connective tissue. It consists of the skeleton, skin, and vascular walls.
If we look in more detail at the mechanism of the disease, we need to start with the fact that each cell of the body that has a nucleus contains 23 pairs of chromosomes. Absolutely each of the chromosomes was formed from 1 molecule of deoxyribonucleic acid (or DNA). DNA contains a large number of genes. Thus, each chromosome contains from 429 to 3511 genes.
One of the genes, fibrillin-1, is located on the long arm of chromosome 15 and is designated FBN1. It is responsible for encoding a large structural protein, which is part of microfibrils (organelles of muscle fibers) and is responsible for creating tissue elasticity and the formation of a strong cytoskeleton (the “skeleton” of cellular structures). Defects in fibrillin-1 lead to mutation.
Elastin fibrils are an integral part of large vessels and ligaments. Often, when these fibrils are disrupted, aneurysms appear.
The syndrome causes damage to transforming growth factor beta (TGF-β), which controls proliferation (proliferation of cellular elements) and cellular differentiation (distribution of functions). There is a disruption in the binding of the inactive form of TGF-β, which will entail an increase in the activity of the factor, which means that all the signs of the syndrome will appear.
Due to a genetic disorder, there is a loss of elasticity of the skin and other connective tissue, and a loss of their strength. Loose joints occur, and the skin can stretch greatly.
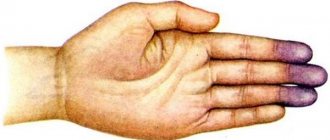
Due to changes in collagen fibers, normal hemostasis fails, which is designed to maintain the liquid state of the blood, dissolve blood clots, and stop bleeding through blood clotting. Defects lead to impaired aggregation (combination) of platelets (one of the elements of blood). Fibrils (cytoplasmic fibers) are involved in hemostasis. Along with slow blood flow inside the vessels, platelet adhesion to elastin occurs (their adherence) with the help of fibronectin. Fibronectin ensures the restoration of structures, creating the necessary components of connective tissue - fibroblasts. Individuals suffering from the syndrome have a deficiency of fibronectin.
Problems with the gastrointestinal tract are caused by an increased content of collagen in the digestive system. There is dysfunction of the biliary tract (bile tract, gall bladder), chronic gastroduodenitis, dolichosigma (abnormal lengthening of one of the sections of the colon), hiatal hernia.
Due to the above factors, characteristic signs of the syndrome arise.
How are they treated?
Since the disease is genetic in nature, and today medicine does not have the tools to correct gene mutations, treatment of Marfan syndrome is aimed at improving the patient’s condition, stopping the progression of the disease and eliminating clinical manifestations. This is a complex process in which different specialists take part, depending on the nature of the most pronounced symptoms - an orthopedist, a cardiologist, an ophthalmologist, a therapist, and doctors of other specialties.
Clinical recommendations for Marfan syndrome include limiting physical activity to the minimum acceptable level to avoid the development of pathologies of the heart and blood vessels, pneumothorax and other dangerous conditions. Treatment efforts include:
- taking medications;
- if necessary, surgical interventions to correct the most damaged areas of the heart and circulatory system;
- orthopedic correction;
- sanatorium treatment, physiotherapy, physical therapy.
If medical recommendations are followed, the prognosis is almost always favorable: through the efforts of doctors, the course of the disease is significantly improved, they get the opportunity to live a long life without serious health complications.
Classification
Marfan syndrome occurs differently for everyone. Considering the severity, it happens:
- light;
- average
; - heavy.
Severe ones are very rare, 1 in 50,000.
Based on how many body structures are affected, Marfan syndrome is classified into 2 forms:
- erased
(there are mild deviations in 1 or 2 systems); - pronounced
(there are implicit deviations in 3 systems or expressed in 1; pronounced deviations in 2, 3 systems or more).
The nature of the course is progressive
(rapidly progressing signs, deterioration of the condition) and
stable
(unchanging signs of the syndrome over many years).
All cases of the disease are divided as follows:
- family
(inherited from generation to generation) – make up 75%; - random
or
sporadic
(disease occurring for the first time in the family) – account for 25%.
Why does the genetic syndrome appear?
The cause of the development of the disease is considered to be a mutation in the FBN1 gene, which is located on chromosome 15 and is responsible for the normal production of fibrillin 1. This protein of connective tissue is one of the main components that gives it elasticity and ability to contract.
The first to be affected by the genetic syndrome are the structures containing the largest amount of important protein - the walls of blood vessels, the ligamentous apparatus, and the ligament of cinnamon of the eye. The altered connective tissue is not able to perform its function or withstand physical stress due to loss of strength and elasticity, and the child develops symptoms of the disease.
The disease is genetic and is transmitted from parents in an autosomal dominant manner. The risk of having a baby with a hereditary syndrome is very high if mom or dad has signs of the disease. In 75% of cases of disease, the appearance of the disease in each generation of the family can be traced. In 25% of patients, a new, spontaneous mutation is detected; there is no clear connection with inheritance.
Connective tissue does not form a separate organ in the human body. But its cells are located throughout the body. By means of these structures, supporting, protective and trophic functions are performed, a kind of framework and integument of all organs are formed. Types of connective tissue include cartilage, bone, muscle, adipose tissue, blood and lymph. Therefore, systemic diseases associated with tissue pathology have a wide variety of manifestations.
Treatment of spider finger disease
Therapy for spider finger syndrome is carried out comprehensively. Main treatment methods:
- The use of drugs that regulate collagen levels. Medicines promote the production of the component in the required quantity and eliminate its deficiency.
- Improving material metabolism. The doctor prescribes large quantities of ascorbic acid and B vitamins.
When it comes to the treatment of Marfan disease, antioxidative drugs, drugs based on anabolic steroids and a small amount of antiplatelet agents are used. In addition to drug treatment, the doctor prescribes a diet that limits the consumption of animal protein.
When a patient suffers from astigmatism, he needs to wear glasses or contacts. This will allow your eyes not to strain and prevent subsequent deterioration of vision.
If the normal shape of the sternum is disrupted with the formation of a funnel, thoracoplasty is indicated. The goal of the procedure is to stop the reduction of the area behind the ribs to avoid injury and compression of the internal systems.
Heart pathologies are treated with surgery. The condition of the aorta is normalized by plastic surgery, and prostheses are installed instead of valves.
Therapeutic gymnastics is mandatory. Your doctor will help you choose a set of exercises. First, exercise therapy is performed under the supervision of a doctor, then the patient can do exercises at home. Physical education helps improve muscle tone and flexibility of the spine.
To prevent the progression of the disease, they resort to rest in sanatoriums and resorts.
Consequences and complications
As the syndrome progresses, the likelihood of complications occurring is quite high. The most common complications are:
- cataract (clouding of the lens);
- glaucoma (periodic increase in intraocular pressure);
- complete blindness;
- postural disorders (latest degrees of kyphosis, scoliosis);
- mitral valve prolapse (sagging of its valves when the left ventricle of the heart contracts);
- congestive heart failure caused by impaired contractility of the heart muscle;
- dissecting aneurysm;
- spontaneous pneumothorax;
- pneumonia;
- stroke;
- thrombosis.
The most serious complication is rupture of the aneurysm, which leads to death.
The most common complication of the syndrome, which ends in death
Family history
Boundaries of the heart in children: methods of determination, norms by age, deviations
In cases of family history, when there are confirmed cases of the same disease in 1st degree relatives (parents, children, siblings), and also when a genetic study has confirmed a mutation of the gene responsible for the synthesis of fibrillin, 1 major clinical criterion is sufficient and involvement of one more system in the pathological process. The reliability of the diagnosis increases if 2 relatives have a large criterion for skeletal damage.
If there are no diagnosed cases of Marfan syndrome in the family, it is necessary to have at least 2 major clinical criteria and damage to the third system in the body.
Forecast
Treatment methods for atherosclerosis of the abdominal aorta and causes of the disease
The average life expectancy for Marfan syndrome is 30-45 years.
It is known that many famous personalities suffered from this syndrome. This is Hans Christian Andersen, a Danish writer, author of the famous Little Mermaid; Abraham Lincoln - 16th President of the United States, Michael Phelps - famous swimmer, multiple Olympic champion. As well as famous composers - Niccolo Paganini, Sergei Rachmaninov.
People with this pathology should carefully monitor their health, constantly monitor and consult with their doctor, and avoid excessive physical activity.
In addition to drug treatment, it is necessary to carry out preventive measures in order to improve general well-being, increase immunity, and an appropriate work and rest schedule.
Pathological anatomy
On morphological examination, the elastic fibers are thinned, unevenly located, and in some places chaotically; There is a dissection of the middle tunic of large vessels, loosening of the endothelial layer, and the formation of cushion-like projections into the lumen of the vessel in the endothelial and subendothelial layers (see Dissecting aneurysm). The elastic framework of the aorta and pulmonary trunk is poorly developed. Myocardium with dystrophic changes, vacuolization, and in places with sharp swelling of fibers. In the bone tissue, there is sparseness of the bone beams and uneven deposition of lime; the structure of cartilaginous tissue is disrupted due to the formation of collagen bundles that stratify the interstitial substance.
Historical reference
The first mention of an unusual disease can be found in the works of the American ophthalmologist E. Williams, who in 1875 described signs of identical displacement of the lenses of the eyes of his siblings. In addition to ophthalmological problems, these children had increased joint mobility and high growth.
The disease gained fame later, 20 years later, when the French pediatrician Antoine Marfan presented his observations of a 5-year-old patient. The little patient was distinguished by unusual skeletal abnormalities and rapid progression of the disease. The syndrome was named after the French doctor, although it later became known that the girl he observed suffered from another hereditary pathology - congenital contractural arachnodactyly.
You can find many examples of the syndrome being detected in talented, famous people. It is believed that violinist Niccolo Paganini, American President Abraham Lincoln, Russian composer Sergei Rachmaninov and other famous personalities suffered from this disease. Some researchers believe that the unusual nature of people with Marfan syndrome is explained by the increased concentration of adrenaline in the blood. This hormone causes an increase in activity and the development of extraordinary abilities.
Prevention
No specific prevention has been developed, so there is no way to prevent the mutation, which in half of the cases will definitely be inherited, but for those already suffering from Marfan syndrome there are a number of restrictions that will prevent them from serious consequences.
Prohibited
:
- strong physical activity;
- participation in sports competitions, scuba diving, contact sports;
- work in hazardous industries;
- being in a place where background radiation is high.
If possible, it is better to change the hot climate to a temperate one.
Patients need to be seen regularly by a doctor (every six months). Light physical activity, race walking, ball games, and basic exercises (squats, turns, stretching) are allowed.
You should not ignore caries, the common cold or helminthic infestations, as they also significantly aggravate the course of the syndrome, since the immunity with such a diagnosis is much weaker than in healthy people.
To prevent Marfan disease, diagnosis should be carried out at the stage of conception in order to calculate all the risks, since the pathology is inherited with a high degree of probability.