Porphyrias are a group of hereditary diseases that occur due to a defect in the formation of heme, resulting in the accumulation of porphyrins or their toxic precursors in the body. Heme is the iron-containing part of hemoglobin, a complex protein “responsible” for binding oxygen, transporting it to tissues and removing carbon dioxide.
During the biosynthesis of heme, compounds called porphyrins are formed. Strictly speaking, heme is a porphyrin, in the center of which there is an iron molecule Fe2+. All porphyrins are red.
Due to a defect in enzyme systems in porphyria, certain pathological metabolic products accumulate in the body. Clinical manifestations of the pathology vary depending on the specific defective gene.
Various types of porphyria occur with a frequency of 7–12 cases per 100 thousand population. Asymptomatic carriage of various genes that can lead to this pathology is approximately 50–100 people per 100 thousand1.
General information
Porphyria disease (syn. porphyrin disease) is a large group of metabolic diseases caused primarily by a hereditary defect in the heme biosynthesis system (the combination of iron ions with porphyrin derivatives) and the accumulation of its toxic metabolites (porphobilinogen/δ-aminolevulinic acid) in the body.
Most porphyrias are congenital diseases with autosomal dominant inheritance. Much less often, porphyrias caused by metabolic disorders are acquired and develop under the influence of various factors that contribute to the process of inhibition of heme synthesis enzymes. The human body undergoes constant endogenous synthesis of purines / pyrimidines . Purines include xanthine , adenine , hypoxanthine , guanine ; to pyrimidines - cytosine , uracil , thymine , orotic acid . They are necessary for storage, transcription/translation of genetic information, cell division/growth, signal transmission and energy storage.
Porphyrins in the human body are synthesized in the cells of the bone marrow, liver, tissues of the nervous system, pancreas and are in both a free and bound state, forming various complex protein compounds with iron ions ( hemoglobin , myoglobin , cytochrome , peroxidase , chromoprotein ) or complexes with sodium, potassium, copper, vanadium, nickel, tin, zinc, manganese, cobalt. At the same time, the mechanism of porphyrin synthesis is the same in the cells of all tissues, but the rate of their formation/duration of existence varies significantly. The main function of porphyrite complexes is their participation in complex metabolic processes (oxygen transport, biological oxidation, photosynthesis, etc.). The end product of purine metabolism is uric acid. Porphyrins are excreted from the body in urine, feces and bile.
Almost any nosological form of porphyria is realized due to a decrease in the activity of enzymes in the heme biosynthesis cycle (inhibition of the heme synthesis process), which is caused by a mutation in the pathognomonic gene and leads to the accumulation of intermediate toxic metabolites. However, this genetic disease does not always manifest itself with severe symptoms, since the pathology does not always manifest itself even when enzyme activity decreases to 50% of normal. And only 18-20% of genetic carriers have characteristic clinical symptoms.
In general, porphyrin disease is a fairly rare disease: the generalized incidence rate for various forms of the disease is 1:20,000 (Wikipedia). At the same time, incidence rates per 100 thousand population vary widely - hereditary coproporphyria: 3–5 cases; intermittent acute intermittent porphyria: within 5–10 cases; variant porphyria – 2–3 cases; porphyria cutanea tarda – 15–20 cases per 100 thousand population. Porphyrias are not endemic diseases (that is, characteristic of a particular area) and occur among the population of different countries with the same frequency.
The disease porphyria occurs both chronically and in the form of acute attacks. The age of onset of the disease also varies significantly: erythropoietic porphyrias manifest mainly at 3-5 years of age (preschool childhood), acute porphyrias develop at 14-16 years of age (period/after puberty), the acquired sporadic form of hepatic cutaneous porphyria occurs in persons after 40 years of age. Due to the vastness of the topic, we will consider only some forms of porphyria , in particular Gunther's disease .
Vampire disease
The connection between these two phenomena: disease and ancient beliefs about bloodsucking people was first stated by Dr. Lee Illis from Great Britain. In 1963, he presented a monograph on Porphyria and the Etiology of Werewolves to the Royal Society of Medicine. The scientist's work contained a detailed comparative analysis of surviving historical evidence that described vampires and the symptoms of porphyria. It turned out that the clinical picture of a rare disease exactly copies the portrait of the most colorful ghoul.
In advanced forms of porphyria, the skin around the lips and gums of patients dries out, causing the incisors to be exposed to the gums, creating the impression of a grin. In addition, a special substance, porphyrin, is deposited on the teeth themselves, which colors a person’s smile (or rather, grin) in a reddish-brown color. The skin on the face and body of such people becomes thinner and bursts from exposure to sunlight, becoming covered with scars and ulcers. The disease also damages cartilage, as well as the organs of which they are composed (primarily the nose and ears). Fingers become crooked. Sunlight gives the poor fellows the most severe torment, because it is under the influence of ultraviolet radiation that the breakdown of hemoglobin begins. Therefore, during the day, people suffering from porphyria try not to appear on the street, and are active only at dusk, closer to night. Either from the torment they experience, or from forced seclusion, or from some internal processes occurring in the body, these people also suffer from neuropsychic disorders and inappropriate, including aggressive behavior.
One can imagine the horror of those who one evening or night by moonlight met one of these “cute guys” on a narrow path. Here you will believe not only in vampires and werewolves, but in anything!
Pathogenesis
The pathogenesis of all forms of porphyria has common links, regardless of the characteristics of the clinical course/tissue affiliation. They are based on the absence/reduction of activity of a certain enzyme in the general chain of heme biosynthesis, which leads to excessive accumulation of products in toxic concentrations of porphyrin metabolism in front of the link where the defective enzyme is localized. Enzyme genes are localized on different chromosomes and have no group linkage.
The process of heme synthesis includes eight successive steps, and each enzyme responds to its own step and is encoded by a specific gene. Accordingly, each form of porphyria has its own specific enzymatic defect. In chronic forms of porphyria, accumulation of coproporphyrin , protoporphyrin and uroporphyrin ; in acute forms - porphobilinogen / DALK (delta-aminolevulinic acid).
How dangerous is the disease and is porphyria completely curable?
Porphyria disease, like all enzymopathies, is dangerous due to its latent period. In the presence of provoking factors, it quickly progresses to a severe course and unforeseen complications.
Currently, medicine does not have radical technologies for curing porphyria. Removal of the spleen has been shown to provide little benefit due to increased red blood cell lifespan and decreased sun sensitivity. Beta-carotene is prescribed. The best effect is provided by protection from exposure to sunlight.
Classification
The qualification of porphyrias is based on such signs as the location of the disturbance in porphyrin metabolism/accumulation of porphyrins and clinical manifestations, according to which the following are distinguished:
- Based on the location of the disturbance in the metabolism of porphyrins, they are distinguished: erythropoietic (primary disorder in the bone marrow) and hepatic porphyrias (disorders develop primarily in the liver);
- According to clinical manifestations, porphyrias are divided into acute forms, manifested by predominantly damage to the nervous system, and forms, manifested by skin damage.
Accordingly, each of the forms includes several types of porphyrias. A generalized classification of forms and types of porphyrias is given in the table below.
What is the connection between porphyria and vampires
According to medical scientists, people believed to be vampires were susceptible to a disease called porphyria, or else they had a rare genetic blood disorder. Porphyria, translated from the Greek "porphyros", means purple and is caused by consanguineous marriages, which was facilitated by low migration, especially in small villages and towns. The inhabitants of the villages of Transylvania were especially susceptible to porphyria about a thousand years ago, however, according to available information, this unusual disease did not escape the royal families.
The symptoms of porphyria have been known since time immemorial, and over time, the disease has received scientific justification for the existence of vampires - their habitats, together with their typical lifestyle and appearance, clearly indicate that the so-called vampires are simply people suffering from porphyria - about vampires and porphyria disease.
Causes
The cause of porphyria in the vast majority of cases is mutations in genes that reduce the activity of the enzyme involved in the process of heme biosynthesis. The disease is inherited predominantly in an autosomal dominant manner, less often in an autosomal recessive manner (Gunther's disease). And only porphyria cutanea tarda (urocoproporphyria) can develop both as a result of prolonged intoxication with heavy metals/liver diseases (liver tumors, alcoholic/viral hepatitis C), and develops in the presence of a hereditary predisposition.
However, for the development of some forms of the disease, in particular acute porphyria , in addition to the presence of a pathological mutation in the enzyme gene, the manifestation of symptoms requires exposure to provoking factors that stimulate the process of porphyrin production. Such generally recognized factors include severe/constant stress, prolonged insolation, alcohol abuse, fasting, infections of bacterial/viral origin, intoxication with heavy metals (mercury, lead), medications (sedatives, NSAIDs, sulfonamides, oral contraceptives, cephalosporins, antimalarial drugs , barbiturates, etc.), in women - menstrual cycle / pregnancy .
The presence of these factors promotes either increased consumption of heme as an end product or stimulation of the activity of the initial enzyme of the biosynthesis cycle. As a result, the synthesis/accumulation of intermediate metabolic products of porphyrins in toxic concentrations is accelerated.
Diagnostic measures
Diagnosis of porphyria is performed by a hematologist. The doctor examines the child or adult and includes in the history all the symptoms that may indicate disturbances in the process of heme synthesis. The patient will have to answer questions about medications taken, diet, and previous infectious diseases. The doctor asks the girls about the stability of the menstrual cycle, pregnancies and abortions.
The next stage of diagnosis is laboratory tests. Patients are prescribed:
- general clinical and biochemical blood tests;
- Ehrlich test;
- study of enzyme concentrations in the blood;
- PCR tests for hepatitis;
- molecular genetic research.
If there are appropriate indications, the child or adult attends a consultation with a dermatologist, nephrologist and gastroenterologist. Differential diagnosis allows doctors to exclude neurological and psychiatric pathologies from the patient’s medical history.
Symptoms
Clinical manifestations of porphyria vary widely and are determined by the specific form of the disease. Let's look at just a few forms of the disease.
Gunther's disease (congenital erythropoietic porphyria)
Like all other erythropoietic porphyrias, Gunther's disease is quite rare. The disease manifests itself in early childhood (aged 1-5 years) and is caused by congenital enzymopathies. The site of porphyrin metabolism disruption is bone marrow erythroblasts. It is registered in families exclusively among sisters/brothers of the same generation and is inherited from either parent as an autosomal recessive trait. At the same time, parents of sick children do not have any clinical/biochemical signs of the disease. Patients with erythropoietic porphyria are characterized by a deficiency of uroporphyrinogen cosynthetase, which is concentrated in the pathological population of erythroblasts and, as a result of such enzymatic blockade, heme biosynthesis is disrupted, which leads to the accumulation of a critical concentration of uroporphyrin I in the child’s body. They occur equally often in males and females.
Gunther's disease is characterized by six main symptoms:
- Increased sensitivity to solar radiation, light with the appearance of blisters on open areas of the skin.
- Absence of typical symptoms for acute porphyria (abdominal symptoms).
- Excessive hair development.
- Discharge of red-colored urine.
- Pinkish-brown color of teeth.
- Enlarged spleen due to hemolytic anemia .
The disease can manifest itself either by all signs or by some of them. Changes in the skin occur mainly in early spring against the background of decreased diuresis and general weakness. Changes appear as itching/redness of the skin on the back of the hands/feet, ears, face, legs and forearms after sun exposure, in place of which bullous elements (vesicles) with serous-hemorrhagic contents later appear. At the same time, the skin is extremely sensitive to mechanical influences. In cases of secondary infection, ulceration of the blisters occurs and healing occurs with the formation of scars.
The skin is uneven, the hands have a claw-like appearance, the nail plates are thickened/deformed/clouded; in some cases, during a chronic course, the auricles are destroyed, and mutilation of the phalanges of the hand is noted with disruption of its function. X-ray shows osteoporosis of the epiphyses of the phalanges , joint contracture (partial/complete) in the distal limbs. Patients are characterized by long eyelashes/thick eyebrows, poor physical development, general emaciation, hyperpigmentation, pale skin, stained teeth, hypertrichosis on the face, and an enlarged spleen. Less commonly, clouding of the lens/cornea. Laboratory tests show a high content of uro/copro/protoporphyrin in feces/urine, plasma and erythrocytes.
Porphyria cutanea tarda
The most common liver porphyria, caused by a disruption in the synthesis of liver hemes, in which there is increased formation/excretion of coproporphyrin/uroporphyrin in the urine and their retention in the skin. Urocoproporphyria manifests itself with three main dermatological symptoms: pigmentation; bubbles; hypertrichosis.
Hyperpigmentation of the skin is diffuse in nature and occurs mainly in areas of the body exposed to solar insolation (hands, neck, face, ears, upper chest). The color varies from reddish-bluish (bronze) to earthy gray and is determined by individual characteristics, the presence of concomitant diseases and the direct influence of external climatic/occupational factors. This characteristic reaction occurs only at the sites of irradiation, and it is regarded as photosensitization (photodermatosis), which is eliminated in the absence of irradiation. The reaction intensifies in summer and practically disappears in winter. Over time, pigmentation becomes persistent and becomes more intense, changing slightly throughout the year.
The next classic sign is a blistering reaction , which is preceded by increased vulnerability of the skin on the back of the hands/face. It is manifested by the formation of blisters on apparently unchanged skin, ranging in size from a millet grain to a round/oval pea, with liquid contents (initially serous, and then becomes cloudy and turns purulent). Subsequently, the blisters spontaneously open, forming erosions with irregular outlines and subsequently forming atrophic scars. The bubble reaction increases in the spring and summer, and in winter there are usually no bubbles. The predominant localization of blisters is the skin of the face, neck, ears, and back of the hands; less often - on the scalp, forearms, lips. In women, there may be an atypical location of blisters - on closed areas of the skin: thighs, back, legs. The duration of the vesicular reaction varies from 1-2 weeks to several months. In cases of secondary infection, lymphadenitis / lymphangitis and deep ulcers . Hypertrichosis is a less specific syndrome, occurring on average in 70% of patients on the face in the frontotemporal region.
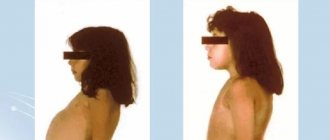
This form is characterized by obesity , dysfunction of the gastrointestinal tract, damage to the liver, the organ of vision: impaired color vision, clouding of the cornea, conjunctivitis , dilatation of the fundus vessels. In addition to these main symptoms, premature skin aging and the appearance of deep wrinkles are characteristic. Nails also undergo changes: they become deformed, lose their shine, and become matte; Subungual hyperkeratosis often develops . Patients look older than their age. Below are photos of patients with porphyria (late cutaneous form).
In acute porphyria, severe abdominal pain, stool retention, increased heart rate, increased blood pressure, and change in urine color (from pink to red-brown) develop. The severity of the patient's condition is mainly due to neurological symptoms - pain throughout the body, decreased sensitivity, progressive muscle weakness, sometimes reaching complete paralysis, convulsive seizures, various mental disorders (anxiety, psychomotor agitation, delirium, hallucinations).
Acute porphyrias: acute intermittent porphyria
It occurs much more often in females. Provoking factors play a significant role in the development of the disease. This form of the disease is characterized by several groups of symptoms:
- Pain syndrome - intense abdominal pain, accompanied by nausea/vomiting, stool disorders (diarrhea/constipation); the pain is paroxysmal in nature, less often - constant and can last from 5-6 hours to several days with localization in various parts of the abdomen.
- Neurological symptoms manifest as flaccid paralysis ; paresis ; polyneuritis ; sensory/bulbar disorders, dysfunction of the urethral sphincter.
- Mental disorders (emotional lability, chronic insomnia , depression / anxiety , psychomotor agitation, delirium, auditory/visual hallucinations, delirium ).
- Hypothalamic dysfunction ( fever of central origin, hyponatremia ).
- Epileptiform seizures with a high risk of developing during acute attacks of coma.
- Cardiovascular system disorders - hypertension , sinus tachycardia , ECG changes.
- Pigmenturia (pink/red urine, dark diffuse skin coloring, chloasma, freckles).
Clinical symptoms and their severity are determined by the form of the disease. Thus, with a latent/healthy carriage of the mutant allele, clinical signs are absent against the background of the presence of minimal biochemical deviations from the norm.
- The latent form is characterized by muscle weakness, periodic abdominal pain, sinus tachycardia , insomnia , hypertension , and, less commonly, psychological personality changes.
- The manifest acute form of intermittent porphyria can occur in several ways. The mild form is characterized by short, periodically occurring acute attacks of the disease, limited to abdominal symptoms, which in most cases end favorably. Severe form : characterized by severe attacks lasting from 2 to 10 weeks with relapses after a few months/1-2 years. Manifests with abdominal symptoms, mental disorders/neurological disorders. May be fatal. Stepped form : it is characterized by an increase in symptoms and deep general disorders with each new attack. Relapses become more frequent and occur within 1-2 months. In most cases, the outcome is unfavorable and every 3-4 attacks are fatal. Acute form : extremely severe course, accompanied by severe general disorders; It is more common during pregnancy and 24-60% of cases end in death from paralysis of the respiratory center.
Porphyria
Porphyrias are a group of metabolic diseases caused by disturbances (usually of a genetic nature) in the activity of specific heme biosynthetic enzymes, leading to oversynthesis and accumulation of intermediate products. Intermediates that accumulate include porphyrins and the porphyrin precursors delta-aminolevulinic acid and porphobilinogen and their derivatives. The clinical picture of the disease manifests itself at toxic concentrations. Patterns of these substances in plasma, red blood cells, urine and feces are characteristic of each porphyria and are the basis for screening tests and more complete biochemical characterization of the disease [3].
Heme biosynthesis involves 8 enzymatic steps in the conversion of glycine and succinyl coenzyme to heme. The first and last 3 enzymes in the pathway are found in the mitochondrion, while the other 4 are found in the cytosol (Figure 1).
Picture 1.
They are encoded by 9 genes, because The first enzyme, delta-aminolevulinate synthase, has 2 genes that encode unique liver (housekeeping) and bone marrow isoenzymes, ALAS1 and ALAS2, respectively. All these genes have been cloned, and mutations in them have been identified (Table 1).
Table 1.
Negative feedback is an important part of the regulation of heme biosynthesis in the liver. Heme, with the help of a repressor protein, binds to DNA, which leads to suppression of transcription and cessation of the synthesis of aminolevulinate synthase (ALAS1), the rate-limiting enzyme in heme biosynthesis. In the liver, an increase in the activity of aminolevulinate synthase is caused by compounds that enhance the microsomal oxidation system (fat-soluble toxins, sulfonamides, steroids, NSAIDs, barbiturates) - while the consumption of heme increases for the formation of cytochrome P450, which reduces the intracellular concentration of free heme. As a result, derepression of enzyme synthesis occurs. As for the bone marrow, regulation here is carried out with the help of iron ions. Iron, by binding to a special iron-binding protein, initiates the synthesis of the bone marrow isoenzyme ALAS2, that is, it has a positive effect (Fig. 2).
Figure 2.
Porphyrias are classified as erythropoietic or hepatic depending on the site of synthesis and accumulation of heme metabolic intermediates.
Erythropoietic porphyrias include congenital erythropoietic porphyria (Gunther's porphyria), erythropoietic protoporphyria and X-linked protoporphyria, which are widespread, especially among children. Hepatic porphyrias include acute porphyrias (of which there are 4), which cause neurological symptoms, usually in the form of acute attacks, and porphyria cutanea tarda, which is the most common of the porphyrias. Acute porphyrias include aminolevulinic acid dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, and variegated porphyria. All forms of acute porphyria are inherited in an autosomal dominant manner and have low (about 10%) penetrance, indicating that a mutation in at least one heme biosynthetic enzyme is important but not sufficient to cause clinical disease. It is necessary to involve some additional genetic factors that are necessary for the pathological phenotype in heterozygous carriers of the mutant gene [1,3].
It is an autosomal recessive genetic disorder characterized by markedly reduced enzymatic activity of uroporphyrogen III synthase, the fourth enzyme in the heme biosynthetic pathway. Defects in uros, the gene encoding this enzyme, are extremely heterogeneous at the molecular level. At least 36 different mutations of this gene are known, as well as one mutation of GATA-1 (encoding an erythroid transcription factor). The disease is characterized by hypersynthesis of uroporphyrin I, which manifests itself at birth and in the neonatal period. Uroporphyrinogen synthase catalyzes the conversion of hydroxymethylbilane (HMB), a linear tetrapyrrole, to a cyclic tetrapyrrole, uroporphyrinogen III. In the absence of this enzyme, HMB undergoes spontaneous ring closure to uroporphyrinogen I, which can only be further metabolized to coproporphyrinogen I. They are spontaneously oxidized to uroporphyrin and coproporphyrin, which are photosensitizing porphyrins and cause the cutaneous manifestations of this pathology [4].
The clinical picture of the disease may vary depending on the severity of inherited uros mutations and the level of porphyrins in plasma and red blood cells. Severe disease may manifest itself in utero (non-immune hydrops fetalis) or shortly after birth with red urine, severe photosensitivity, hemolytic anemia, splenomegaly and erythrodontia (Fig. 3). Repeated exposure to sunlight can lead to scarring and infection. Skeletal changes may result from vitamin D deficiency. The characteristic subepidermal bullous lesions progress to form cortical erosions that heal with scarring and areas of hyper- and hypopigmentation. Hypertrichosis and alopecia are also common. Loss of facial features and fingers are common and are the result of recurrent blisters, infections, and scarring [3,4].
Figure 3.
Diagnosis is often made after birth, when severe photosensitivity and red urine discoloration begin. To confirm the diagnosis, DNA is examined to identify causative mutations. This is especially important for genetic counseling and for prenatal diagnosis in subsequent pregnancies. Gunther's disease should be differentiated from other skin lesions (solar erythema, trauma, allergic, infectious lesions, etc.). For this, various biochemical tests and fluorescence spectroscopy are used (Table 2).
Treatment begins with sun protection. Care should be taken to avoid skin injury; and early treatment of skin infections is warranted. Long-term transfusion therapy eliminates anemia and effectively suppresses erythropoiesis. However, they can lead to iron excess, which may require parenteral deferoxamine or the oral iron chelators deferasirox or deferiprone. More invasive procedures such as splenectomy have been aimed at reducing hemolysis, but long-term effectiveness remains questionable. Hematopoietic stem cell transplantation is the treatment of choice when a suitable donor is available, especially for younger patients. With successful transplantation, there is a marked improvement in the patient's condition and a decrease in porphyrin levels, even if they do not completely normalize [3,4].
EPP is also an inherited disorder characterized by the accumulation of protoporphyrin in the blood, red blood cells, and tissues, resulting in painful photosensitivity. It is reported to have a worldwide prevalence of 1:75,000 to 1:200,000. An important feature of EPP is that it is widespread among children.
Classical EPP results from inheritance of a mutant fech allele encoding a ferrochelatase that catalyzes the incorporation of iron into the protoporphyrin ring to produce heme. Ferrochelatase also catalyzes the incorporation of zinc into this ring to form zinc protoporphyrin from any protoporphyrin that remains after heme synthesis is complete. In EPP, metal-free protoporphyrin accumulates in bone marrow reticulocytes because the formation of both heme and zinc protoporphyrin is impaired. Metal-free protoporphyrin enters the plasma from the bone marrow and circulating red blood cells and is then transported to the skin, causing photosensitivity, and to the liver for excretion in the bile. More than 130 identified mutations in the gene, located on chromosome 18, are loss-of-function mutations. More recently, it was discovered that 5–10% of patients with a typical EPP phenotype have an X-linked mutant Alas2 gene, and this variant of EPP is now called X-linked protoporphyria [4].
Accumulation of lipid-soluble free protoporphyrin in the skin and cutaneous blood vessels and subsequent photoactivation by sunlight leads to the characteristic cutaneous manifestations of EPP. Children with this disorder develop pain, redness, swelling and itching within minutes of exposure to sunlight. These symptoms can last from several hours to several days. Unlike Gunther's disease, vesicles and bullae are uncommon and occur in only 10% of cases. Chronic skin changes such as lichenification and pseudovesicles may develop after repeated episodes of photosensitivity. They are most noticeable in the cheekbone area and hands. Various hepatobiliary complications may also develop. Obstruction of the bile ducts by gallstones, which contain protoporphyrin, is observed in 20% of patients. In 3–5% of patients, decreased bile outflow can lead to pigmentary cirrhosis.
Moderate hypochromic anemia with microcytosis is observed. The level of serum iron and soluble transferrin receptors does not change. Neurovisceral manifestations may occur in patients with complicated EPP (hepatopathy) who develop motor neuropathies similar to those observed in acute porphyrias.
Biochemical tests are performed to determine the level of protoporphyrin in plasma and red blood cells because there is a marked increase in protoporphyrin levels in EPP. Because protoporphyrin is not excreted in the urine, urinary levels of porphyrin precursors are normal. To confirm the diagnosis, analysis of the fech and Alas2 genes is possible. Additionally, hepatobiliary function is examined for prognostic purposes.
As with Gunther's disease, sun protection is an integral part of the management of patients with EPP. Patients are prescribed an alpha-melanocyte-stimulating hormone analogue (afamelanotide). In the presence of complications from the liver, bile acid sequestrants (cholestyramine 4–16 g/day) and choleretic drugs—urodeoxycholic acid—are recommended.
This is a very rare autosomal recessive disorder caused by a severe deficiency of aminolevulinate dehydratase (porphobilinogen synthase). This enzyme catalyzes the formation of porphobilinogen from two molecules of aminolevulinate. The enzyme is encoded by the alad gene, located on chromosome 9q34. Full enzyme activity requires a sulfhydryl group and one zinc atom per subunit (8 identical subunits in total). Due to enzyme deficiency, aminolevulinate levels are significantly elevated in plasma and urine in patients with this disease. ALA is further metabolized to form other heme precursors, and as a result, coproporphyrin III is excreted in the urine in large quantities, and the amount of protoporphyrin IX increases in red blood cells. It should be said that, as in other acute porphyrias, the mechanism of neurological damage is not well understood [3,4].
To date, only 6 cases of this disease have been reported. In 4 cases, symptoms manifested only in adolescence. The patients had neurovisceral symptoms and no skin manifestations, which is a characteristic feature of acute porphyrias.
Laboratory diagnostics reveal insufficient activity of the defective enzyme and a marked increase in the level of coproporphyrin III in the urine.
Differential diagnosis should include other hepatic porphyrias, lead poisoning, hereditary tyrosinemia type 1, in which the activity of aminolevulinate dehydratase is suppressed.
Because only a few cases have been reported, treatment recommendations are based on limited experience. Currently, hemin is used, which to some extent improves the clinical picture. Using a negative feedback mechanism, it eliminates heme deficiency, suppresses the activity of delta-aminolevulinic acid synthase, a key enzyme in the synthesis of porphyrins, as a result of which the synthesis of porphyrins and toxic metabolic precursors of heme decreases. Thus, hemin corrects the pathophysiological disorders accompanying porphyria.
Acute intermittent porphyria (Swedish porphyria, pyrroloporphyria), inherited in an autosomal dominant manner, is the most common of the acute porphyrias worldwide, with an estimated prevalence of approximately 5:100,000 in the United States, 1–2:100,000 in Europe. The disease was first described by the Dutch physician Stokwis in 1889, who concluded that his patient's symptoms were caused by a barbiturate (sulfonal). Waldenström in 1937 noted the high prevalence of the disease in the area of northern Sweden and described the disease in more detail. In Sweden, the prevalence of pyrroloporphyria is approximately 4 times higher than in other parts of Europe due to a founder effect centered in Lapland [3,4].
AKI develops in individuals who are heterozygous for mutations in porfibilinogen deaminase (PBHD, the third enzyme in the heme biosynthetic pathway). It catalyzes the conversion of four molecules of porphobilinogen into hydroxymethylbilane, a linear tetrapyrrole. Partial deficiency of PBHD rarely causes clinical symptoms of Swedish porphyria, and most people who inherit a mutant gene for this enzyme remain healthy throughout their lives. Certain drugs and hormones (sulfonamides, barbiturates, anticonvulsants, NSAIDs, estrogen) can directly induce the activity of aminolevulinate synthase, as well as CYP enzymes. This leads to an increase in heme synthesis, as the need for it has increased. When heme synthesis is stimulated, PBHD deficiency becomes apparent because heme synthesis is interrupted midway. Negative feedback induces the work of the rate-limiting enzyme, delta-aminolevulinate synthase, which leads to oversynthesis of aminolevulinate and porphobilinogen, which accumulate in toxic concentrations and cause the clinical picture of the disease.
Most often, the disease manifests itself after reaching puberty. Among the patients, females predominate, which is associated with the functioning of their reproductive system. The clinical picture of AKI is dominated by damage to the nervous system, caused by an excess of aminolevulinate and porphobilinogen in tissues, leading to segmental demyelination of nerve fibers with disruption of their conductivity. The course of the disease is intermittent, there are acute attacks of abdominal pain without peritoneal signs, which are accompanied by dyspepsia, tachycardia, mental disorders (hallucinations, anxiety, insomnia), convulsions, peripheral neuropathy (can progress to respiratory paralysis) and the release of red/brown urine. Attacks are triggered by fasting, steroid hormones or their metabolites, certain medications, and stress. Bulbar disorders - dysphonia, dysphagia, dysarthria - are characteristic of advanced cases. Seizures may be associated with hyponatremia resulting from damage to the hypothalamus and excessively high ADH levels. Other causes of hyponatremia are increased renal sodium loss and decreased gastrointestinal absorption. Convulsions in this case are bad because a number of anticonvulsants are porphyrinogenic and can aggravate the severity of the disease. In addition, porphyrin precursors have a cytotoxic and vasospastic effect on the nephron, which leads to renal failure [1,3,4].
Clinical improvement after liver transplantation in patients with severe AKI clearly indicates that the liver plays an important role in the neuropathic processes of acute porphyrias. The mechanism of neurological dysfunction in acute porphyrias is not clear, and the following hypotheses exist [3]:
- Neurotoxicity of heme biosynthesis intermediates is the basis of neurological dysfunction. This hypothesis is the most preferred, although the evidence is inconclusive.
- PBHD deficiency in nervous system tissues may limit heme synthesis and the formation of important hemoproteins. For example, decreased NO synthase activity may reduce NO production and cause vasospasm, which explains some of the cerebral manifestations of AKI. However, the regulation of heme and hemoprotein synthesis in neural tissue and blood vessels is difficult to study, and conclusive evidence is currently lacking.
- Impaired hepatic heme synthesis during acute attacks may result in decreased tryptophan pyrrolase activity, which may increase plasma and brain tryptophan levels, leading to increased serotonin synthesis and accumulation of neurotoxic doses.
The diagnosis of acute porphyria is verified by quantitative determination of porphyrins and their precursors in the urine. Acute attacks are characterized by a high content of total porphyrins and their precursors: aminolevulinate and porphobilinogen in the urine. In addition, high levels of the 2 heme precursors listed above often remain high during latency, which is a hallmark of AKI when compared with other forms of acute porphyrias. Based on the combination of characteristic clinical signs with an increased content of total porphyrins and their precursors in the urine, feces and blood, as well as with a reduced activity of PBHD in erythrocytes, it is possible to establish the correct diagnosis of patients with AKI. For asymptomatic carriers, differential biochemical diagnosis is difficult. Measuring PBHD activity in erythrocytes does not always provide a clear answer to the question of the presence of the disease, since the ranges of enzyme activity levels in such patients overlap with normal values. In this case, the only way to clarify the diagnosis is a molecular genetic study to identify a mutation in the PBHD gene (Fig. 4: differential diagnosis scheme).
Figure 4.
If the diagnosis is established, potentially dangerous medications should be avoided to prevent acute attacks. In women, attacks may be associated with the luteal phase of the menstrual cycle. To suppress ovulation, they are prescribed gonadotropin-releasing hormone (GHRH) analogues. Due to the use of GnRH analogues, a decrease in bone tissue density is possible. Bisphosphonates are prescribed to prevent fractures. In addition, long-term monitoring of liver function, kidney function and blood pressure levels is recommended for patients over 50 years of age.
In the event of an attack, it is necessary to immediately begin relief of severe symptoms, infusion therapy and correction of electrolyte disturbances, and monitoring of vital functions. If possible, eliminate provoking factors and begin symptomatic therapy:
- analgesics - to relieve pain;
- short-acting benzodiazepines in low doses - to eliminate mental disorders;
- chlorpromazine - to relieve nausea and vomiting;
- beta blockers - to control hypertension and tachycardia.
Relatively safe anticonvulsants include gabapentin, vigabatrin and clonazepam. In severe cases, intravenous infusion of hemin is indicated, which, through negative feedback, suppresses the transcription of hepatic aminolevulinate synthase (ALAS1). This reduces the formation of porphyrin precursors.
Hereditary coproporphyria is inherited in an autosomal dominant manner and often occurs latently due to low penetrance. NCP is the least common among acute porphyrias. This disease is characterized by impaired activity of coproporphyrinogen oxidase, a mitochondrial enzyme that converts coproporphyrinogen III into protoporphyrinogen IX. When enzyme activity is impaired, coproporphyrinogen III accumulates and is excreted in feces and urine. NCP clinical manifestations resemble acute intermittent porphyria.
NCP is an acute hepatic porphyria with severe neurovisceral symptoms. Acute attacks are often associated with the use of certain drugs, fasting and menstruation. Attacks usually begin with mild abdominal pain that slowly gets worse over hours or days, often accompanied by nausea and vomiting. Usually the pain is poorly localized, but in some cases it mimics acute inflammation of the gallbladder, appendix or other intra-abdominal organ. Sometimes the pain predominates in the back or limbs. Motor neuropathy can develop over days or weeks if left untreated. Respiratory failure may be a consequence of impaired innervation of the diaphragm and respiratory muscles. Skin manifestations are very rare. It should be said that patients with any type of acute porphyria, if they are heterozygous for the corresponding mutant genes, may develop chronic changes in the kidneys and liver, which are often subclinical. One of the manifestations of these changes in the liver is hepatocellular carcinoma, which occurs mainly after 60 years of age [3,4].
The diagnosis is made after quantitative analysis of porphyrins in both urine and feces. A threefold increase in porphobilinogen and coproporphyrin is detected in the urine. Molecular genetic tests are used to confirm the diagnosis. Differential diagnosis should include other hepatic porphyrias, lead poisoning, Rotor syndrome (hereditary pigmentary hepatosis) and nonspecific coproporphyrinuria, which also have increased levels of porphyrin precursors [2] (Fig. 4).
Acute attacks are treated by stopping any medications thought to cause them. Then symptomatic and infusion therapy is started, as for AKI.
The disease is called "variegated" because it can cause severe neurovisceral symptoms or photodermatosis identical to those seen in porphyria cutanea tarda. VP is also referred to in the literature as mixed or South African genetic porphyria. VP is an autosomal dominant genetic disorder due to deficient activity of the mitochondrial enzyme protoporphyrinogen oxidase (PPOX). The prevalence of VP is 1:300 in the white population of Dutch origin in South Africa due to the founder effect. A lower prevalence of CAP has been estimated in Finland (1.3:100,000) and Europe as a whole (0.3:100,000) [2].
Clinical manifestations are more common in women than in men. There are acute attacks identical to those found in AKI. Characteristic symptoms include abdominal, chest and limb pain, hypertension, tachycardia, restlessness, seizures and neuromuscular weakness, which can progress to quadriplegia and respiratory paralysis. Phototoxic manifestations may include subepidermal vesicles, bullae, erosions, or ulcers that are slow to heal.
The diagnosis of CAP is established by biochemical testing and confirmed by the identification of a heterozygous mutation in ppox (Table 2). The management of patients with acute attacks is the same as for AKI. Sun should be avoided as there is no effective treatment for skin manifestations. It is important to distinguish CAP from porphyria cutanea tarda because the skin manifestations are very similar [4].
Table 2.
PKP is a hepatic porphyria that manifests as photodermatosis and has no neurological signs. Therefore, this disease is different from the acute hepatic porphyrias discussed above. Since PKP is the most common porphyria, ranging from 1:5000 to 1:70,000 in different countries, it is important to distinguish it from other porphyrias that have cutaneous manifestations. The disease usually manifests in middle/old age and is therefore called late. It is also known by many other names, including symptomatic porphyria, idiosyncratic porphyria, chemical porphyria, or acquired hepatic porphyria [4,5].
This is the only porphyria that can develop in the absence of an inherited mutation of the affected enzyme. Only 20% of patients with this disease have a heterozygous uroporphyrinogen decarboxylase mutation, which reduces uroporphyrinogen decarboxylase activity in all tissues to half of normal. These mutations increase susceptibility to the development of PEP, but additional factors must be present. All this leads to the fact that the enzyme activity decreases by 20% of the normal level and the disease manifests itself. Thus, PKD is a heterogeneous disorder in which environmental, infectious, and genetic factors interact to cause significant deficiency of enzyme activity in the liver. The disease has 3 forms depending on the presence of mutations in the urod gene [3,5]:
- sporadic form. The most common form of porphyria cutanea tarda without the urod mutation;
- family form. Patients are heterozygous for mutations of the urod gene, there is a partial decrease in enzyme activity, and it is transmitted in an autosomal dominant manner. Now some authors call it hepatoerythropoietic porphyria. It manifests in childhood not only with severe skin manifestations, but also with hemolytic anemia, to some extent resembling VEP (Fig. 5);
- acquired form. Apparently, there is a genetic predisposition that leads to a decrease in enzyme activity in hepatocytes. It is believed that as yet unidentified genes other than urod are involved in the development of this form [3,4].
Figure 5.
In the pathogenesis of PKD, the formation of a uroporphyrinogen decarboxylase inhibitor, which is formed after the oxidation of uroporphyrinogen or hydroxymethylbilane, plays an important role. This process requires the presence of high levels of iron, CYP-4501A2 activity and estrogen. But the exact structure and nature of the inhibitor remain unknown. Iron overload and long-term estrogen intake are additional factors causing the manifestation of latent PKD through unknown mechanisms. This is evidenced by the data that more than half of patients have hemosiderosis and elevated levels of plasma ferritin. In addition, removal of iron from the body returns enzyme activity to normal levels in sporadic disease [4].
The main risk factors for PEP are excessive alcohol consumption, smoking, iron overload, hepatitis C virus, HIV and estrogen use (oral contraceptives, prostate cancer treatment). One or more of these factors can be found in more than 80% of patients. Genetic predisposition is an important factor, but it alone is not sufficient for the development of PPC; other factors are also necessary [5].
There are several proposed mechanisms by which alcohol leads to decreased uroporphyrinogen decarboxylase activity. Alcohol increases iron absorption, which leads to iron accumulation in the liver, activates hepatic ALAS-1, free radical formation, and is hepatotoxic. Alcohol also inhibits other enzymes in the heme synthesis pathway. Alcohol, along with the other factors listed above, causes enzyme deficiency, which leads to the accumulation of porphyrins in the liver [5].
Hepatitis C virus and HIV infection have been shown to be associated with PEP. The role of hepatitis C is not entirely clear. A possible hypothesis is that it causes clinical disease in genetically susceptible individuals. As for HIV infection, it somehow affects heme biosynthesis and disrupts the functioning of the cytochrome P-450 system.
Uroporphyrinogen decarboxylase catalyzes the stepwise decarboxylation of uroporphyrinogen to coproporphyrinogen. A decrease in enzyme activity leads to an increase in the production of uroporphyrins. These compounds are automatically oxidized to porphyrins, which accumulate in the liver in large quantities and are transported through the blood to the skin, where they act as photosensitizers. They interact with light with a wavelength of about 400 nm; when activated, they generate reactive oxygen species that cause skin damage - bullae, milia (Fig. 6). Chronic damage to the skin can lead to scarring and changes in skin pigmentation. Other cutaneous manifestations may include periorbital heliotrope, chloracne, hypertrichosis, alopecia, and onycholysis [5].
Figure 6.
Patients' urine is pink-red in color. The diagnosis is made by finding a significant increase in the amount of porphyrins in the urine or plasma with a predominance of carboxylated porphyrins (uroporphyrin and hepta-, hexa- and pentacarboxyporphyrins). Coproporphyrins also increase. Urinary levels of delta-aminolevulinic acid and porphobilinogen are normal or slightly elevated. Typically, individuals with PEP have slightly elevated serum aminotransferase levels and gamma-glutamyl transpeptidase levels are also elevated. Nonspecific results from routine laboratory tests make it clear that special tests are needed to diagnose PKP. High sensitivity of fluometric methods. The latter help to distinguish PPP from other hepatic porphyrias (hereditary coproporphyria, variegated porphyria), which also have cutaneous manifestations [4,5].
As with other types of cutaneous porphyria, avoiding sun exposure is the most effective way to prevent skin manifestations. Therapy targets the pathogenic mechanism of PEP and includes iron reduction, antimalarials that remove porphyrins from the liver and other tissues, and phlebotomy. Patients are first advised to stop smoking, drinking alcohol and taking estrogen. In addition, it is recommended to follow a diet to prevent a compensatory increase in iron absorption in the gastrointestinal tract. Phlebotomy is a fairly safe and effective treatment method. It consists of withdrawing 400–500 ml of blood weekly or even less frequently until the serum ferritin level is <25 μg/L. Clinical remission usually occurs after removal of 2–4 liters of blood. Skin manifestations gradually improve with resolution of the bullae within 2–3 months, and complete biochemical remission is observed after 13 months. During phlebotomy, care must be taken to prevent hemoglobin levels from falling below 110 g/L. Pre-existing anemia and severe serum protein deficiency (eg, due to cirrhosis and bleeding) are relative contraindications to phlebotomy [5].
If phlebotomy is contraindicated, low-dose chloroquine (125 mg every other day) is the next treatment of choice. Chloroquine binds carboxyporphyrins, which accumulate in lysosomes, and, by forming water-soluble complexes with them, it leads to an increase in their excretion in urine. However, it is hepatotoxic. Subcutaneous administration of the iron chelator deferoxamine is also an alternative to phlebotomy. Although not as inexpensive as therapeutic phlebotomy, deferoxamine can remove toxic iron implicated in the pathogenesis of the disease. In addition, it may reduce the compensatory increase in intestinal iron absorption often seen in remission caused by the greater degree of iron reduction achieved by phlebotomies. Oral iron chelators (deferasirox, deferiprone) are available in some countries. They may also be effective and relatively safe for the treatment of PEP [5].
Sources:
- Pustovoit Ya. S. Federal clinical guidelines for the diagnosis and treatment of acute porphyrias. – 2013.
- DiMauro S. et al. Gene Reviews //University of Washington: Seattle, WA, USA. – 2010.
- Kaushansky K., Lichtman MA Williams Hematology. – 2021. – P. 2528.
- Ramanujam VMS, Anderson KE Porphyria diagnostics—part 1: a brief overview of the porphyrias //Current protocols in human genetics. – 2015. – P. 17.20. 1-17.20. 26.
- Ryan Caballes F., Sendi H., Bonkovsky HL Hepatitis C, porphyria cutanea tarda and liver iron: an update // Liver International. – 2012. – T. 32. – No. 6. – pp. 880-893.
Tests and diagnostics
For diagnostic purposes, special laboratory methods are used for the determination of porphyrins/their precursors in feces and urine, and in the blood to assess enzyme activity.
Thus, for Gunther's disease , clinical and biochemical blood tests are characterized by signs of hemolysis - increased serum iron/indirect bilirubin, poikilocytosis , reticulocytosis , spherocytosis , anisocytosis , increased liver transaminases . Patients with acute porphyria are characterized by a decrease in sodium/glucose levels; in cases of late cutaneous porphyria, an increase in ferritin/serum iron and positive markers for the hepatitis C virus.
When making a diagnosis, family history is important: the presence of the disease in close relatives, as well as the circumstances of the onset of the disease (pregnancy, menstruation, insolation, alcohol/medicine intake, fasting, infections, etc.). To confirm the diagnosis, the levels of various enzymes of the heme biosynthesis cycle in plasma, erythrocytes, and lymphocytes are determined.
Among the instrumental methods, CT/ultrasound of the abdominal organs, CT/radiography of the chest, ECG, EEG of the brain, etc. can be prescribed.
Clinical case
Patient T
., 20 years old, on December 23, 2017, during training in martial arts, he received a blow to the stomach, after which he noted an episode of nausea. After 4 days, severe pain in the epigastric region, nausea, and vomiting of bile appeared. He did not receive treatment; within 2 days the symptoms regressed. On 01/05/18, I again felt pain in the epigastric region, accompanied by vomiting and loose stools, but my body temperature did not increase. After 2 days, he was hospitalized at the district hospital at his place of residence with a diagnosis of acute pancreatitis. He denied previous illnesses and requests for medical help. Antisecretory therapy was prescribed and fasting was recommended. Over the course of 2 weeks, he lost 13 kg of body weight and developed weakness and pain in the extremities. On January 16, 2018, the patient was transferred to the intensive care unit (ICU) of the regional hospital. The examination revealed electrolyte disturbances (sodium level - 128 mmol/l, potassium - 2.8 mmol/l), amylasemia (amylase level - 223 IU/l), stenosis of the celiac trunk (80% according to computed tomographic angiography). Conservative therapy did not lead to an improvement in the patient’s condition, and therefore on January 28, 2018 he was hospitalized at the Federal Scientific and Clinical Center for Specialized Medical Care and Medical Technologies with a diagnosis of “Celiac stenosis. Chronic abdominal ischemia syndrome. Cachexia. Electrolyte disturbances" to resolve the issue of surgical treatment. Upon admission: the patient's condition is serious. The skin is of normal color and normal moisture. Body temperature 37.7 °C. Breathing is spontaneous, vesicular, with a frequency of 18 per minute, blood hemoglobin saturation with oxygen is 98%. Blood pressure - 155/110 mm Hg, heart rate - 115 beats/min. According to electrocardiography - sinus tachycardia. The abdomen was not distended, soft and painless on palpation, there were no symptoms of peritoneal irritation. He absorbed enteral nutrition, and there was a decrease in appetite and constipation for 1 week. He did not control urination; the color of the urine was straw-yellow. The patient was consulted by a vascular surgeon regarding stenosis of the celiac trunk; it was decided to refrain from surgical tactics, and conservative treatment was recommended. When examined by a neurologist: consciousness is clear, Glasgow Coma Scale (GCS) - 15 points. Orientation in place, time, and self is complete. Meningeal symptoms are negative. Pupils OD = OS, reaction to light is normal. Full movement of the eyeballs. The face is symmetrical, the tongue is in the midline. There was no dysarthria, dysphonia, or dysphagia. Muscle tone is reduced. Tetraparesis according to the muscle strength rating scale: in the arms up to 2 points in the proximal and up to 4 points in the distal sections, in the legs - up to 3 points in the proximal and distal sections. Tendon reflexes were not evoked. There were no sensory disturbances. There were pelvic function disorders such as urinary incontinence and constipation. The neurologist recommended further examination (electroneuromyography (ENMG), magnetic resonance imaging (MRI) of the brain, cervical and thoracic spine, examination of the cerebrospinal fluid) for differential diagnosis and clarification of the genesis of polyneuropathy. Laboratory examination revealed leukocytosis, mild anemia and thrombocytopenia (leukocytes 13.3 109/l, hemoglobin 12.3 g/dl, platelets 114 109/l, respectively), increased levels of pancreatic amylase (77 U/l), hypoalbuminemia (30 g/l), electrolyte disturbances (sodium 124 mmol/l, potassium 3.5 mmol/l), increased levels of inflammatory markers (C-reactive protein 47.6 mg/l, procalcitonin 7.4 ng/ml), prolongation of activated partial thromboplastin time (40 s), decrease in serum concentrations of iron (2.2 µmol/l) and folic acid (1.9 ng/ml). Computed tomography (CT) of the chest organs revealed single lesions in the lower lobes of the lungs.
Correction of water and electrolyte disturbances (infusions of sodium chloride solutions 0.9% and potassium chloride 4%) and anemia (folic acid, ferrous sulfate), empirical antimicrobial therapy (levofloxacin), mixed nutrition, prevention of stress ulcers (omeprazole), prophylaxis began thrombotic complications (enoxaparin sodium), rehabilitation measures in the ICU, including mechanotherapy in bedside simulators, physical therapy (physical therapy), placement in a bedside chair.
As part of the additional examination, ENMG revealed axonal demyelinating (primarily axonal) motor-sensory polyneuropathy. MRI revealed no evidence of stenosis of the cervical and thoracic spinal canals, the presence of space-occupying formations, or focal brain damage. Analysis of cerebrospinal fluid dated January 30, 2018: colorless, transparent, cytosis 2 cells/μl, glucose 3.9 mmol/l, protein 0.2 g/l; virological data (Epstein-Barr virus, cytomegalovirus, herpes simplex virus types 1 and 2) are negative. A bacteriological examination of the blood, the distal end of the removed venous catheter and sputum revealed the growth of Staphylococcus aureus, and therefore, as of January 30, 2018, levofloxacin was replaced by linezolid.
Taking into account the results of additional examination, the patient’s condition was assessed as a manifestation of Guillain-Barré syndrome and cachexia with nutritional deficiency due to pancreatitis, complicated by the development of nosocomial pneumonia, as well as catheter-associated bloodstream infection and thrombosis of the jugular and subclavian veins. On January 31, 2018, a 5-day course of specific therapy for Guillain-Barré syndrome with immunoglobulin G was started - the drug Octagam (Octapharma Pharmaceutica Production GmbH, Austria) at a dose of 0.4 mg per 1 kg of body weight per day.
Despite the treatment and daily rehabilitation measures, neurological symptoms remained at the same level. 02/05/18 pain in the limbs intensified. Considering the long history of immobilization in the ICU, the symptoms were regarded as manifestations of PTS, and fluvoxamine and carbamazepine were added to therapy. In addition, electrolyte disturbances persisted, and in order to exclude adrenal insufficiency, the patient was further examined: daily urinary sodium excretion was 87 mmol/l, serum cortisol level was 19.7 μg/dl. Taking into account the data obtained, hydrocortisone was added to therapy. Due to the ineffectiveness of linezolid monotherapy (re-growth of Staphylococcus aureus in the blood on 02/05/18, persistent high body temperature), antimicrobial therapy was intensified with cefazolin. Repeated duplex scanning revealed persistent thrombosis of the subclavian and internal jugular veins, which was probably the cause of persistent bacteremia despite antimicrobial therapy.
To exclude infective endocarditis, transesophageal echocardiography was performed, in which no evidence was obtained for the presence of vegetations on the valve apparatus and in the cavities of the heart. During esophagogastroduodenoscopy, fungal esophagitis was discovered, which was later confirmed by microscopy results.
After starting therapy with carbamazepine and fluconazole on 02/08/18, the patient’s condition showed negative dynamics in the form of a decrease in the level of consciousness to stupor (14 points on the GCS), the appearance of spontaneous nystagmus, and nausea. Repeated MRI of the brain to exclude pontine myelinolysis revealed no changes. The next day, clear consciousness was restored, but the patient became emotionally labile and began to complain of increasing aching pain in the limbs and heart, while there were no changes in the electrocardiogram.
Based on the totality of all clinical, laboratory and instrumental data, the patient was suspected of acute porphyria, which required special laboratory confirmation. On 02/09/18, a urine test revealed porphobilinogen (PBG) 200.6 mg/l, δ-aminolevulinic acid (ALA) 145.6 mg/l. The patient was transferred to the intensive care unit of the National Medical Research Center for Hematology, where he remained for 25 days. As part of the pathogenetic therapy of acute porphyria, a constant infusion of 20% glucose solution in a volume of 1000 ml/day was carried out, and a course of heme arginate (Normosang, Orphan Europe SarL, France) 125 mg/day was administered. Over time, as a result of the therapy, a decrease in the concentration of PBG from 200 to 31 mg/l and ALA from 146 to 23 mg/l was noted. The patient continued antibacterial therapy for staphylococcal infection (focal bilateral pneumonia, bloodstream infection). Pyelonephritis caused by Pseudomonas aeruginosa was diagnosed.
Treatment was carried out for thrombosis of the veins of the lower extremities and the jugular vein on the right. Due to persistent nutritional deficiency, the patient received mixed nutrition. The course of the disease in the intensive care unit was complicated by the development of symptomatic psychosis, and therefore haloperidol therapy was carried out with a positive effect. Laboratory tests revealed signs of folate deficiency and iron deficiency anemia, and folic acid replacement therapy was carried out. With the correction of hyponatremia, it was possible to stabilize the sodium level within 132 mmol/l (the minimum level reached 120 mmol/l). Kinesiotherapy and exercise therapy classes were conducted daily. On 04/04/18 the patient was discharged. His condition was regarded as moderate and was due to the course of the underlying disease, persistent flaccid paresis of the limbs, nutritional deficiency, anemia of combined origin, and thrombotic complications.
Prevention
There is no specific prevention. In order to minimize attacks, it is recommended:
- Avoid/minimize exposure to porphyrinogenic factors (regular lifestyle, avoid stress, taking potentially dangerous medications, drinking alcoholic beverages, fasting/strict diets, sun exposure).
- Patients over 50 years of age should undergo twice-yearly examinations to evaluate liver health.
- Rational employment.
- Carrying out preventive treatment/clinical examination of patients.
- If there is a porphyria patient in the family, it is necessary to carry out DNA diagnostics (detection of genetic mutations) and determine the activity of various enzymes of the heme synthesis cycle.
Consequences and complications
Since porphyrias, with the exception of porphyria cutanea tarda, are hereditary, their cure is completely impossible. If a patient has 2-3 or more attacks per year, a neurological deficit in the form of flaccid peripheral paresis may persist for a long time. In severe cases, complications are possible in the form of bulbar syndrome , hyponatremia , paralysis of the respiratory muscles with a high risk of death. In case of porphyria with skin lesions, there is a risk of developing sclerodermoid processes.
general information
Porphyrin disease is diagnosed relatively rarely: Russian doctors detect no more than 12 cases per 100 thousand people. Various forms of pathology are becoming widespread in certain regions of the Earth. Thus, signs of porphyria cutanea tarda are often detected in residents of South Africa (1 case per 800 people). Acute intermittent type of disease is typical for residents of Northern Europe (1 case per 1000 people). Men and women are equally likely to suffer from various forms of heme synthesis.
List of sources
- Kuznetsova N.P., Pankov B.S., Chubarova A.S. and others. Porphyria. M., 1981. p. 66–14.
- Pustovoit Y.S., Pivnik A.V., Karpova I.V. Clinic, diagnosis and treatment of porphyria // Manual for doctors. Moscow, 2003
- Vorobyov A, Kravchenko S, Kremenetskaya A, Karpova I, Pustovoit Y. Acute intermittent porphyria: problems of diagnosis and treatment // Doctor. 2003 No. 2, pp. 8-13.
- Diagnosis and treatment of acute porphyrias: Clinical recommendations of the national hematological society / ed. Pustovoit Y.S., Kravchenko S.K., Shmakova R.G., Savchenko V.G. — 2021.
- Guide to hematology in 3 volumes / ed. Vorobyova A.I. – 2005 – T.3.