Порфирии — это группа наследственных заболеваний, которые возникают из-за нарушения образования гема, в результате чего в организме накапливаются порфирины или их токсичные предшественники. Гем — железосодержащая часть гемоглобина, сложный белок, «ответственный» за связывание кислорода, транспорт его в ткани и выведение углекислого газа.
В процессе биосинтеза гема образуются соединения под названием порфирины. Строго говоря, гем — это порфирин, в центре которого находится молекула железа Fe2+. Все порфирины имеют красный цвет.
Из-за дефекта ферментных систем при порфириях в организме накапливаются те или иные патологические продукты обмена веществ. Клинические проявления патологии разнятся в зависимости от конкретного дефектного гена.
Различного рода порфирии встречаются с частотой 7–12 случаев на 100 тысяч населения. Бессимптомное носительство различных генов, способных привести к этой патологии, составляет примерно 50–100 человек на 100 тысяч1.
Общие сведения
Болезнь порфирия (син. порфириновая болезнь) представляет собой большую группу заболеваний обмена веществ, обусловленных преимущественно наследственным дефектом в системе биосинтеза гема (соединение ионов железа с производными порфирина) и накоплением в организме его токсичных метаболитов (порфобилиногена/δ-аминолевулиновой кислоты). Большинство порфирий являются врождёнными заболеваниями с наследованием по аутосомно-доминантному типу. Значительно реже порфирии, обусловленные нарушением метаболизма, являются приобретенным и развиваются под воздействием различных факторов, способствующих процессу ингибирования ферментов синтеза гема.
В человеческом организме происходит постоянный эндогенный синтез пуринов/пиримидинов. К пуринам относятся ксантин, аденин, гипоксантин, гуанин; к пиримидинам – цитозин, урацил, тимин, оротовая кислота. Они необходимы для хранения, транскрипции/трансляции генетической информации, деления/роста клеток, передачи сигналов и накопления энергии.
Порфирины в человеческом организме синтезируются в клетках костного мозга, печени, тканях нервной системы, поджелудочной железе и находятся как в свободном, так и связанном состоянии, образуя различные сложные белковые соединения с ионами железа (гемоглобин, миоглобин, цитохром, пероксидазу, хромопротеид) или комплексы с натрием, калием, медью, ванадием, никелем, оловом, цинком, марганцем, кобальтом. При этом, механизм синтеза порфиринов одинаков в клетках всех тканей, однако скорость их образования/длительность существования существенно варьирует. Основной функцией порфиритовых комплексов является их участие в сложных метаболических процессах (транспортировка кислорода, биологическое окисление, фотосинтез и др.). Конечным продуктом пуринового метаболизма является мочевая кислота. Порфирины выделяются из организма с мочой, калом и желчью.
Практически любая нозологическая форма порфирии реализуется вследствие снижения активности ферментов в цикле биосинтеза гема (торможение процесса синтеза гема), что обусловлено мутацией в патогномоничном гене и приводит к накоплению промежуточных токсичных метаболитов. Однако это генетическое заболевание не всегда манифестирует выраженной симптоматикой, поскольку патология не всегда себя проявляет даже при снижении активности фермента до 50% от нормы. И только 18-20% генетических носителей имеют характерную клиническую симптоматику.
В целом порфириновая болезнь относятся к достаточно редким заболеваниям: обобщенный показатель заболеваемости различными формами болезни составляет 1:20 000 (Википедия). При этом, показатели заболеваемости на 100 тыс. населения широко варьирует — наследственная копропорфирия: 3–5 случаев; перемежающаяся острая перемежающаяся порфирия: в пределах 5–10 случаев; вариантная порфирия – 2–3 случая; поздняя кожная порфирия – 15–20 случаев на 100 тыс. населения. Порфирии не эндемичные заболевания (то есть характерные для определенной местности) и встречаются среди населения различных стран с одинаковой частотой.
Заболевание порфирия протекает как хронически, так и в виде острых атак. Существенно варьирует и возраст дебюта заболевания: эритропоэтические порфирии манифестируют преимущественно в 3-5 лет (дошкольном детстве), острые порфирии развиваются в 14-16 лет (период/после полового созревания), приобретенная спорадическая форма печеночной кожной порфирии — у лиц после 40 лет. Ввиду обширности темы рассмотрим лишь некоторые формы порфирий, в частности болезнь Гюнтера.
Болезнь вампиров
О связи двух этих явлений: болезни и древних верований о людях-кровососах впервые заявил доктор Ли Иллис из Великобритании. В 1963 году он представил в Королевское медицинское общество монографию «О порфирии и этиологии оборотней». Труд ученого содержал подробный сравнительный анализ сохранившихся исторических свидетельств, в которых описывались вампиры, и симптомов порфирии. Оказалось, что клиническая картина редкой болезни в точности копирует портрет самого колоритного вурдалака.
При запущенной форме порфирии кожа вокруг губ и десен у больных высыхает, отчего резцы обнажаются до десен, создавая впечатление оскала. К тому же на самих зубах откладывается особое вещество порфирин, которое окрашивает улыбку (а вернее, оскал) человека в красновато-бурый цвет. Кожа на лице и теле таких людей истончается и от воздействия солнечного света лопается, покрываясь шрамами и язвами. Болезнь повреждает еще и хрящи, а также те органы, из которых они состоят (прежде всего, это нос и уши). Пальцы становятся скрюченными. Солнечный свет доставляет бедолагам наиболее тяжкие мучения, ведь именно под воздействием ультрафиолета начинается распад гемоглобина. Поэтому днем люди, страдающие порфирией, стараются на улице не появляться, а активность проявляют только в сумерки, ближе к ночи. То ли от испытываемых мучений, то ли от вынужденного затворничества, то ли от каких-то внутренних процессов, происходящих в организме, эти люди страдают еще и нервно-психическими расстройствами и неадекватным, в том числе, агрессивным поведением.
Можно себе представить ужас тех, кто однажды вечером или ночью при свете луны встретил на узкой дорожке одного из таких «симпатяг». Тут не только в вампиров и оборотней поверишь, а во что угодно!
Патогенез
Патогенез всех форм порфирий имеет общие звенья, независимо от особенностей клинического течения/тканевой принадлежности. В их основе — отсутствие/снижение активности определённого фермента в общей цепи биосинтеза гема, что приводит к избыточному накоплению продукции в токсических концентрациях порфиринового обмена перед звеном, где локализуется дефектный энзим. Локализуются гены ферментов на разных хромосомах и групповой сцепленности не имеют.
Процесс синтезирование гема включает восемь последовательных этапов и каждый фермент отвечает свой этап и кодируется определенным геном. Соответственно каждая форма порфирии имеет свой специфичный ферментативный дефект. При хронических формах порфирий отмечается накопление копропорфирина, протопорфирина и уропорфирина; при острых формах — порфобилиногена/ДАЛК (дельта-аминолевулиновой кислоты).
Чем опасно заболевание и лечится ли порфирия полностью
Болезнь порфирия, как все ферментопатии, опасна своим скрытым периодом. При наличии провоцирующих факторов она быстро переходит к тяжелому течению, непредвиденным осложнениям.
В настоящее время медицина не имеет радикальных технологий излечения порфирии. Показано, что удаление селезенки дает несущественную пользу вследствие увеличения сроков жизни эритроцитов и уменьшает чувствительность к солнцу. Назначают прием бета-каротина. Наилучший эффект дает защита от попадания под солнечные лучи.
Классификация
В основу квалификации порфирий положены такие признаки как место нарушения метаболизма порфиринов/накопления порфиринов и клинические проявления, согласно которым выделяют:
- по месту нарушения метаболизма порфиринов различают: эритропоэтические (первичное нарушение в костном мозге) и печеночные порфирии (нарушения развиваются первично в печени);
- по клиническим проявлениям порфирии делятся на острые формы, проявляющиеся преимущественным поражением нервной системы и формы, манифестирующие поражением кожи.
Соответственно, каждая из форм включает несколько видов порфирий. Обобщенная классификация форм и видов порфирий приведена в таблице ниже.
Какая связь между порфирией и вампирами
Согласно мнению ученых-медиков люди, которых считали вампирами, были подвержены болезни под названием порфирия, а иначе у них было редко встречающееся генетическое заболевание крови. Порфирия, в переводе с греческого «porphyros», означает пурпурный и причиной ее возникновения являются родственные браки, чему способствовала низкая миграция населения, особенно в небольших деревнях и городах. Особенно сильно были подвержены порфирии, жители сел Трансильвании около тысячи лет назад, однако, по имеющимся сведениям, эта необычная болезнь не миновала и королевские семьи.
О симптомах порфирии известно с незапамятных времен, а со временем болезнь получила и научное обоснование существования вампиров – места их обитания в совокупности с типичным образом жизни и внешностью, ясно указывают на то, что так называемые вампиры, это попросту страдающие порфирией люди — о вампирах и болезни порфирия.
Причины
Причиной порфирий в подавляющем большинстве случаев являются мутации в генах, способствующие снижению активности фермента, принимающего участие в процессе биосинтеза гема. Наследуется заболевание преимущественно по аутосомно-доминантному, реже по аутосомно-рецессивному типу (болезнь Гюнтера). И только поздняя кожная порфирия (урокопропорфирия) может развиваться как вследствие длительной интоксикации тяжелыми металлами/заболеваний печени (опухоли печени, алкогольный/вирусный гепатит С), так и развивается при наличии наследственной предрасположенности.
Однако, для развития некоторых форм заболевания, в частности острая порфирия, кроме присутствия патологической мутации в гене фермента для манифестации симптоматики требуется воздействие провоцирующих факторов, которые стимулируют процесс выработки порфиринов. К такими общепризнанным факторам относятся сильные/постоянные стрессы, длительная инсоляция, злоупотребление алкоголем, голодание, инфекции бактериального/вирусного генеза, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства (седативные средства, НПВС, сульфаниламиды, оральные контрацептивы, цефалоспорины, антималярийные препараты, барбитураты и др.), у женщин — менструальный цикл/беременность.
Наличие этих факторов способствует или повышенному потреблению гема, как конечного продукта или стимуляции активности начального фермента цикла биосинтеза. Как следствие — ускоряется синтез/накопление промежуточных продуктов обмена порфиринов в токсических концентрациях.
Диагностические мероприятия
Диагностика порфирии выполняется гематологом. Врач осматривает ребенка или взрослого и включает в анамнез все симптомы, которые могут указывать на нарушения в процессе синтеза гема. Пациенту предстоит ответить на вопросы о принимаемых медикаментах, режиме питания, перенесенных инфекционных заболеваниях. Девушек врач расспрашивает о стабильности менструального цикла, беременностях и абортах.
Следующим этапом диагностики становятся лабораторные тесты. Пациентам назначаются:
- общеклинический и биохимический анализы крови;
- проба Эрлиха;
- исследование концентрации ферментов в крови;
- ПЦР-тесты на гепатит;
- молекулярно-генетические исследования.
При наличии соответствующих показаний ребенок или взрослый посещает консультацию дерматолога, нефролога и гастроэнтеролога. Дифференциальная диагностика позволяет врачам исключить из анамнеза пациента неврологические и психиатрические патологии.
Симптомы
Клинические проявления порфирий варьируют в широких пределах и определяются конкретной формой заболевания. Рассмотрим лишь несколько форм заболевания.
Болезнь Гюнтера (врожденная эритропоэтическая порфирия)
Как и все другие эритропоэтические порфирии болезнь Гюнтера достаточно редкое явление. Манифестирует заболевание уже в раннем детстве (в возрасте 1-5 лет) и обусловлено врожденными ферментопатиями. Местом нарушения порфиринового обмена являются эритробласты костного мозга. Регистрируется в семьях исключительно среди сестер/братьев одного поколения и наследуется от любого из родителей как аутосомно-рецессивное. При этом, у родителей больных детей какие-либо клинические/биохимические признаки болезни отсутствуют. Для больных эритропоэтической порфирией характерен дефицит косинтетазы уропорфириногена, который концентрируется в патологической популяции эритробластов и в результате такой ферментативной блокады нарушается биосинтез гема, что приводит к накоплению в организме ребёнка критической концентрации уропорфирина I. Встречаются одинаково часто у лиц мужского/женского пола.
Для болезни Гюнтера характерны шесть основных признаков:
- Повышенная чувствительность к солнечной радиации, свету с появлением на открытых участках кожи пузырей.
- Отсутствие типичной симптоматики для острой порфирии (абдоминальных симптомов).
- Чрезмерное развитие волосяного покрова.
- Выделение окрашенной в красный цвет мочи.
- Розовато-коричневая окраска зубов.
- Увеличение селезенки на фоне гемолитической анемии.
Манифестировать заболевание может как всеми признаками, так отдельными из них. Изменения со стороны кожи возникают преимущественно ранней весной на фоне сниженного диуреза и общей слабости. Проявляются изменения зудом/покраснением кожи на тыльной стороне кистей/стоп, ушных раковинах, лице, на голени и предплечьях после инсоляции, на месте которых позже появляются буллезные элементы (пузырьки) с серозно-геморрагическим содержимым. При этом кожа чрезвычайно чувствительна к механическим воздействиям. В случаях присоединении вторичной инфекции происходит изъязвление пузырьков и заживление с образованием рубцов.
Кожа неровная, кисти имеют когтеобразный вид, ногтевые пластинки утолщены/деформированы/помутневшие; в ряде случаев при хроническом течении разрушаются ушные раковины, отмечается мутиляция фаланг кисти с нарушением ее функции. На рентгенограмме остеопороз эпифизов фаланг, контрактура суставов (частичная/полная) в дистальных отделах конечностей. Для пациентов характерны длинные ресницы/густые брови, слабое физическое развитие, общее истощение, гиперпигментация, бледность кожи, окрашенные зубы, гипертрихоз на лице, увеличенная селезенка. Реже — помутнение хрусталика/роговицы. Лабораторно в кале/моче, плазме и в эритроцитах отмечается высокое содержание уро/копро/протопорфирина.
Поздняя кожная порфирия
Наиболее часто встречаемая порфирия печени, обусловленная нарушением процесса синтеза гемов печени, при которой отмечается повышенное образование/выделение копропорфирина/уропорфирина с мочой и их задержкой в кожных покровах. Урокопропорфирия манифестирует тремя основными дерматологическими симптомами: пигментация; пузыри; гипертрихоз.
Гиперпигментация кожи носит диффузный характер и возникает преимущественно на участках тела, подвергающихся солнечной инсоляции (кисти рук, шея, лицо, ушные раковины, верхняя часть груди). Цвет варьирует от красновато-синюшного (бронзового) до землисто-серого и определяется индивидуальными особенностями, наличием сопутствующих заболеваний и непосредственным влиянием внешних климатических/профессиональных факторов. Такая характерная реакция возникает лишь на местах облучения, и она расценивают как фотосенсибилизация (фотодерматоз), который при отсутствии облучения устраняется. Реакция усиливается летом, а зимой практически исчезает. По прошествии времени пигментация приобретает стойкий характер и становится более интенсивной, слабо изменяясь на протяжении года.
Следующим классическим признаком является пузырная реакция, которой предшествует повышенная ранимость кожи тыльной стороны кистей/лица. Проявляется образованием на видимо неизмененной коже пузырей размерами от просяного зерна до горошины округлой/овальной формы с жидкостным содержимым (вначале серозным, а затем мутнеет и переходит в гнойное). В дальнейшем пузыри самопроизвольно вскрываются, образуя эрозии с неправильными очертаниями с формированием впоследствии атрофических рубцов. Пузырная реакция увеличивается в весенне-летний период, а зимой пузыри, как правило, отсутствуют. Преимущественной локализацией пузырей являются кожа лица, шеи, ушных раковин, тыла кистей; реже — на волосистой части головы, предплечьях, губах. У женщин может иметь место атипичное расположение пузырей — на закрытых участках кожи: бедрах, спине, голенях. Длительность пузырной реакции варьирует от 1-2 недель до нескольких месяцев. В случаях присоединения вторичной инфекции отмечается лимфаденит/лимфангоит, глубокие язвы. Гипертрихоз – менее специфический синдром, встречается в среднем у 70% пациентов на лице в лобно-височной области.
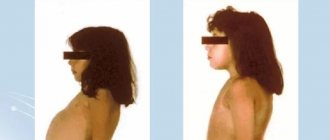
Для этой формы свойственны ожирение, нарушение функций ЖКТ, поражения печени, органа зрения: нарушение цветовосприятия, помутнение роговицы, конъюнктивит, расширение сосудов глазного дна. Кроме этих основных симптомов характерно преждевременное старение кожи и появление глубоких морщины. Ногти также подвергаются изменению: деформируются, теряют блеск, становятся матовыми; часто развивается подногтевой гиперкератоз. Пациенты выглядят старше своего возраста. Ниже приведены фото больных порфирией (поздняя кожная форма).
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
Острые порфирии: острая перемежающаяся порфирия
Встречается значительно чаще у лиц женского пола. В развитии заболевания существенную роль играют провоцирующие факторы. Для этой формы заболевания характерны несколько групп симптомов:
- Болевой синдром — интенсивные боли в животе, сопровождаемые тошнотой/рвотой, расстройствами стула (диарея/запоры); боли носят приступообразный характер, реже – постоянный и могут продолжаться от 5-6 часов до нескольких дней с локализацией в различных отделах живота.
- Неврологическая симптоматика манифестирует вялыми параличами; парезами; полиневритом; сенсорными/бульбарными нарушениями, расстройством функции уретрального сфинктера.
- Психические расстройства (эмоциональная лабильность, хроническая бессонница, депрессии/тревоги, психомоторное возбуждение, бред, слуховые/зрительные галлюцинации, делирий).
- Гипоталамическая дисфункция (лихорадка центрального генеза, гипонатриемии).
- Эпилептиформные припадки с высоким риском развития во время острых приступов коматозного состояния.
- Нарушения со стороны сердечно-сосудистой системы — гипертензия, синусовая тахикардия, изменения на ЭКГ.
- Пигментурия (розовая/красная моча, темная диффузная окраска кожи, хлоазмы, веснушки).
Клиническая симптоматика и ее выраженность определяется формой заболевания. Так, при скрытом/здоровом носительстве мутантного аллеля клинические признаки отсутствуют на фоне присутствия минимальных биохимических отклонений от нормы.
- Для латентной формы характерны мышечная слабость, периодически возникающие боли в животе, синусовая тахикардия, бессонница, гипертензия, реже — психологические изменения личности.
- Манифестные острая форма перемежающейся порфирии может протекать в нескольких вариантах. Лёгкая форма характеризуется непродолжительными периодически возникающими острыми приступами болезни, ограничивающиеся абдоминальными симптомами, которые заканчиваются в большинстве случаев благоприятно. Тяжёлая форма: характерны тяжёлые приступы длительностью от 2 до 10 недель с рецидивами через несколько месяцев/1-2 года. Манифестирует абдоминальными симптомами, психическими расстройствами/неврологическими нарушениями. Может закончиться летально. Ступенчатая форма: для нее характерным является нарастание симптоматики и глубоких общих нарушений с каждым новым приступом. Рецидивы учащаются и наступают через 1-2 месяца. В большинстве случаев исход неблагоприятный и каждый 3-4 приступ заканчивается летально. Острейшая форма: крайне тяжелое течение, сопровождаемое тяжелыми общими нарушениями; чаще встречается во время беременности и 24-60% случаев заканчивается летально от паралича дыхательного центра.
Порфирии
Порфирии являются группой метаболических заболеваний, вызванных нарушениями (обычно генетической природы) в активности конкретных ферментов биосинтеза гема, приводящими к сверхсинтезу и накоплению промежуточных продуктов. Промежуточные продукты, которые накапливаются, включают порфирины и предшественников порфиринов — дельта-аминолевулиновую кислоту и порфобилиногена и их производных. Клиника болезни проявляется при токсических концентрациях. Образцы этих веществ в плазме, эритроцитах, моче и фекалиях характерны для каждой порфирии и являются основой для скрининговых тестов и более полной биохимической характеристики болезни [3].
Биосинтез гема включает 8 ферментативных стадий в превращении глицина и сукцинил-коэнзима в гем. Первый и последние 3 фермента в пути находятся в митохондрии, тогда как другие 4 находятся в цитозоле (рис. 1).
Рисунок 1.
Они кодируются 9 генами, т.к. первый фермент — дельта-аминолевулинатсинтаза имеет 2 гена, которые кодируют уникальные печёночный (housekeeping) и костномозговой изоферменты, ALAS1 и ALAS2 соответственно. Все эти гены были клонированы, и мутации в них были идентифицированы (таб. 1).
Таблица 1.
Обратная негативная связь является важной частью регуляции биосинтеза гема в печени. Гем с помощью белка репрессора связывается с ДНК, что приводит к подавлению транскрипции и прекращению синтеза аминолевулинатсинтазы (ALAS1), скорость-лимитирующего фермента в биосинтезе гема. В печени повышение активности аминолевулинатсинтазы вызывают соединения, усиливающие работу микросомальной системы окисления (жирорастворимые токсины, сульфаниламиды, стероиды, НПВС, барбитураты) — при этом возрастает потребление гема для образования цитохрома Р450, что снижает внутриклеточную концентрацию свободного гема. В результате происходит дерепрессия синтеза фермента. Что касается костного мозга, то здесь регуляция осуществляется с помощью ионов железа. Железо, связываясь с особым железосвязывающим белком, инициирует синтез костномозгового изофермента ALAS2, то есть оказывает положительный эффект (рис. 2).
Рисунок 2.
Порфирии классифицируются как эритропоэтические или печёночные в зависимости от места синтеза и накопления промежуточных продуктов метаболизма гема.
Эритропоэтические порфирии — это врождённая эритропоэтическая порфирия (порфирия Гюнтера), эритропоэтическая протопорфирия и X-сцепленная протопорфирия, которая широко распространена, особенно среди детей. Печёночные порфирии включают острые порфирии (их 4), которые вызывают неврологические симптомы, обычно в виде острых приступов, и позднюю кожную порфирию, которая является наиболее распространённой из порфирий. Острые порфирии включают порфирию, обусловленную дефицитом дегидратазы аминолевулиновой кислоты, острую перемежающуюся порфирию, наследственную копропорфирию и вариегатную порфирию. Все формы острой порфирии наследуются аутосомно-доминантно и имеют низкую (около 10%) пенетрантность, что указывает на то, что мутация по крайней мере в одном ферменте биосинтеза гема важна, но недостаточна для клинического проявления заболевания. Необходимо задействовать некоторые дополнительные генетические факторы, которые необходимы для патологического фенотипа у гетерозиготных носителей мутантного гена [1,3].
Это аутосомно-рецессивное генетическое заболевание, характеризуемое заметно сниженной ферментативной активностью уропорфироген-III-синтазы, четвёртого фермента в пути биосинтеза гема. Дефекты uros, гена, кодирующего этот фермент, являются чрезвычайно гетерогенными на молекулярном уровне. Известны по меньшей мере 36 различных мутаций этого гена, а также одна мутация GATA-1 (кодирует эритроидный фактор транскрипции). Болезнь характеризуется гиперсинтезом уропорфирина I, который проявляется при рождении и в неонатальном периоде. Уропорфирогенсинтаза катализирует превращение гидроксиметилбилана (ГМБ), линейного тетрапиррола, в циклический тетрапиррол — уропорфириноген III. В отсутствие этого фермента ГМБ подвергается спонтанному замыканию кольца до уропорфириногена I, который может быть дополнительно метаболизирован только до копропорфириногена I. Они спонтанно окисляются до уропорфирина и копропорфирина, которые являются фотосенсибилизирующими порфиринами и вызывают кожные проявления этой патологии [4].
Клиника болезни может варьироваться в зависимости от тяжести унаследованных мутаций uros и уровня порфиринов в плазме и эритроцитах. Тяжёлое заболевание может проявлять себя ещё во внутриутробном периоде (неиммунная водянка плода) или вскоре после рождения выделением красной мочи, сильной фоточувствительностью, гемолитической анемией, спленомегалией и эритродонтией (рис. 3). Повторное воздействие солнечного света может привести к образованию рубцов, инфекции. Изменения со стороны скелета могут быть результатом дефицита витамина D. Характерные субэпидермальные буллёзные поражения прогрессируют и образуют корковые эрозии, заживающие с образованием рубцов и участков гипер- и гипопигментации. Также распространённым является гипертрихоз и алопеция. Потеря черт лица и пальцев встречаются часто и являются результатом рецидивирующих пузырей, инфекций и рубцевания [3,4].
Рисунок 3.
Диагноз часто ставится после рождения, когда начинается тяжёлая фотосенсибилизация и окрашивание мочи в красный цвет. Для подтверждения диагноза исследуют ДНК для выявления причинных мутаций. Это особенно важно для генетического консультирования и для пренатальной диагностики в последующей беременности. Болезнь Гюнтера следует дифференцировать от других поражений кожи (солнечная эритема, травмы, аллергические, инфекционные поражения и др.). Для этого используют различные биохимические тесты и флуоресцентную спектроскопию (таб. 2).
Лечение начинается с защитных мер от солнца. Следует соблюдать осторожность, чтобы избежать травм кожи; и раннее лечение кожных инфекций является оправданным. Длительная трансфузионная терапия устраняет анемию и эффективно подавляют эритропоэз. Однако они могут привести к избытку железа, что может потребовать парентерального дефероксамина или орально-активных хелаторов железа, деферазирокса или деферипрона. Более инвазивные процедуры, такие как спленэктомия, были направлены на снижение гемолиза, но долгосрочная эффективность по-прежнему вызывает сомнение. Трансплантация гемопоэтических стволовых клеток — это метод выбора, когда имеется подходящий донор, особенно для молодых пациентов. При успешной трансплантации наблюдается заметное улучшение состояния больного и снижение уровней порфирина, даже если они не полностью нормализуются [3,4].
ЭПП также является наследственным нарушением, характеризующимся накоплением протопорфирина в крови, эритроцитах и тканях, что приводит к болезненной фоточувствительности. Сообщается, что во всём мире она распространена с частотой от 1:75000 до 1:200000. Важной особенностью ЭПП является то, что она отличается широкой распространённостью среди детей.
Классическая ЭПП возникает в результате наследования мутантного аллеля fech, кодирующего феррохелатазу, которая катализирует включение железа в кольцо протопорфирина для получения гема. Феррохелатаза также катализирует включение цинка в это кольцо с образованием цинк-протопорфирина из любого протопорфирина, который остаётся после завершения синтеза гема. При ЭПП безметалловый протопорфирин накапливается в ретикулоцитах костного мозга, поскольку нарушается образование как гема, так и цинк-протопорфирина. Безметалловый протопорфирин поступает в плазму из костного мозга и циркулирующих эритроцитов, а затем переносится на кожу, вызывая фоточувствительность, и в печень для экскреции с желчью. Более 130 идентифицированных мутаций в гене, находящемся в 18-й хромосоме, относятся к мутациям с потерей функций. Совсем недавно было обнаружено, что у 5‒10% пациентов с типичным фенотипом ЭПП Х-сцепленно передаётся мутантный ген Alas2, и сейчас этот вариант ЭПП называют Х-сцепленной протопорфирией [4].
Накопление жирорастворимого свободного протопорфирина в коже и кожных кровеносных сосудах и последующая фотоактивация солнечным светом приводят к характерным кожным проявлениям ЭПП. У детей с этим расстройством развиваются боль, покраснение, отёк и зуд в течение нескольких минут после воздействия солнечного света. Эти симптомы могут длиться от нескольких часов до нескольких дней. В отличие от болезни Гюнтера, везикулы и буллы нехарактерны, встречаются только в 10% случаев. Хронические изменения кожи, такие как лихенизация и псевдовезикулы, могут развиться после повторных эпизодов фоточувствительности. Они наиболее заметны в скуловой области и кисти рук. Также могут развиться различные гепатобилиарные осложнения. Обструкция жёлчевыводящих путей жёлчными камнями, в состав которых входит протопорфирин, наблюдается у 20% пациентов. У 3‒5% пациентов снижение оттока желчи может привести к пигментному циррозу.
Наблюдается умеренная гипохромная анемия с микроцитозом. Уровень сывороточного железа и растворимых рецепторов трансферрина не изменяется. Нейровисцеральные проявления могут быть у пациентов с осложнённой ЭПП (гепатопатия), у которых развиваются двигательные нейропатии, аналогичные той, которая наблюдалась в острых порфириях.
Проводят биохимические тесты для определения уровня протопорфирина в плазме и эритроцитах, потому что при ЭПП наблюдается заметное повышение уровня протопорфирина. Поскольку протопорфирин не выводится с мочой, уровни предшественников порфирина в моче являются нормальными. Для подтверждения диагноза возможен анализ генов fech и Alas2. Дополнительно исследуют гепатобилиарную функцию в прогностических целях.
Как и при болезни Гюнтера, защитные меры от солнца являются неотъемлемой частью ведения пациентов с ЭПП. Пациентам назначается аналог альфа-меланоцитстимулирующего гормона (афамеланотид). При наличии осложнений со стороны печени рекомендуют секвестранты жёлчных кислот (холестирамин по 4‒16 г/сут) и желчегонные — уродезоксихолевую кислоту.
Это очень редкое аутосомно-рецессивное заболевание, вызванное серьёзным дефицитом аминолевулинатдегидратазы (порфобилиногенсинтазы). Этот фермент катализирует образование порфобилиногена из двух молекул аминолевулината. Фермент кодируется геном alad, локализованным на хромосоме 9q34. Для полной активности фермента требуется сульфгидрильная группа и один атом цинка на субъединицу (всего 8 одинаковых субъединиц). Из-за дефицита фермента уровень аминолевулината существенно повышается в плазме и моче у пациентов с этой болезнью. ALA дополнительно метаболизируется с образованием других предшественников гема, и в результате копропорфирин III выделяется с мочой в больших количествах, а количество протопорфирина IX увеличивается в эритроцитах. Следует сказать, что, как и в других острых порфириях, механизм неврологического повреждения недостаточно изучен [3,4].
На сегодняшний день зарегистрировано всего 6 случаев этой болезни. В 4 случаях симптомы манифестировали только в подростковом периоде. У больных наблюдались нейровисцеральные симптомы и отсутствовали кожные проявления, что является характерной особенностью острых порфирий.
При лабораторной диагностике определяется недостаточная активность дефектного фермента, выраженное повышение уровня копропорфирина III в моче.
Дифференциальная диагностика должна включать другие печёночные порфирии, отравление свинцом, наследственную тирозинемию 1 типа, в которых подавляется активность аминолевулинатдегидратазы.
Поскольку было зарегистрировано всего несколько случаев, рекомендации по лечению основаны на ограниченном опыте. На данный момент используется гемин, который в какой-то степени улучшает клиническую картину. Он по механизму отрицательной обратной связи устраняет дефицит гема, подавляет активность синтазы дельта-аминолевулиновой кислоты — ключевого фермента синтеза порфиринов, в результате чего уменьшается синтез порфиринов и токсичных метаболических предшественников гема. Таким образом гемин осуществляет коррекцию патофизиологических нарушений, сопровождающих порфирию.
Острая перемежающаяся порфирия (шведская порфирия, пирролопорфирия), наследуемая аутосомно-доминантно, является наиболее распространённой из острых порфирий во всем мире с предполагаемой распространённостью приблизительно 5:100000 в Соединенных Штатах, 1‒2:100000 в Европе. Болезнь была впервые описана голландским врачом Стоквисом в 1889 г., который пришел к выводу, что симптомы у его пациента были вызваны барбитуратом (сульфоналом). Вальденстрём в 1937 г. отметил высокую распространённость болезни в районе северной Швеции и более подробно описал болезнь. В Швеции распространенность пирролопорфирии примерно в 4 раза выше, чем в других частях Европы из-за эффекта основателя, сосредоточенного в Лапландии [3,4].
ОПП развивается у лиц, которые являются гетерозиготными по мутациям порфибилиногендезаминазы (ПБГД, третий фермент в пути биосинтеза гема). Она катализирует превращение четырёх молекул порфобилиногена в гидроксиметилбилан, в линейный тетрапиррол. Частичный дефицит ПБГД редко вызывает клиническую картину шведской порфирии, и большинство людей, которые наследуют мутантный ген этого фермента, остаются здоровыми на протяжении всей жизни. Определённые препараты и гормоны (сульфаниламиды, барбитураты, противосудорожные, НПВС, эстроген) могут непосредственно индуцировать работу аминолевулинатсинтазы, а также ферментов CYP. Это приводит к увеличению синтеза гема, поскольку повысилась потребность в нём. Когда стимулируется синтез гема, дефицит ПБГД даёт о себе знать, потому что синтез гема прерывается в середине пути. По отрицательной обратной связи индуцируется работа скорость-лимитирующего фермента — дельта-аминолевулинатсинтазы, что приводит к сверхсинтезу аминолевулината и порфобилиногена, которые накапливаются в токсических концентрациях и вызывают клиническую картину болезни.
Чаще всего заболевание манифестирует после достижения пубертатного возраста. Среди заболевших преобладают лица женского пола, что связано с функционированием их репродуктивной системы. В клинике ОПП доминирует поражение нервной системы, обусловленное избытком аминолевулината и порфобилиногена в тканях, приводящих к сегментарной демиелинизации нервных волокон с нарушением их проводимости. Течение болезни носит интермиттирующий характер, имеются острые приступы абдоминальных болей без перитонеальных признаков, которые сопровождаются диспепсией, тахикардией, психическими расстройствами (галлюцинации, беспокойство, инсомния), судорогами, периферической нейропатией (может прогрессировать до респираторного паралича) и выделением красной/бурой мочи. Приступы провоцируются голоданием, стероидными гормонами или их метаболитами, некоторыми лекарствами и стрессом. Бульбарные нарушения — дисфония, дисфагия, дизартрия — характерны для запущенных случаев. Судороги могут быть связаны с гипонатриемией, возникающей в результате поражения гипоталамуса и чрезмерно высоких уровней АДГ. Другие причины гипонатриемии — увеличенная потеря натрия почками и снижение абсорбции в ЖКТ. Судороги в данном случае плохи тем, что ряд противосудорожных препаратов являются порфириногенными и могут усугублять тяжесть заболевания. Кроме этого, предшественники порфирина имеют цитотоксическое и вазоспастическое воздействие на нефрон, что приводит к почечной недостаточности [1,3,4].
Клиническое улучшение после трансплантации печени у пациентов с тяжёлым ОПП чётко указывает на то, что печень играет важную роль в нейропатических процессах острых порфирий. Механизм неврологической дисфункции в острых порфириях не ясен, и имеются следующие гипотезы [3]:
- Нейротоксичность промежуточных продуктов биосинтеза гема является основой неврологической дисфункции. Эта гипотеза наиболее предпочтительна, хотя доказательства не убедительны.
- Дефицит ПБГД в тканях нервной системы может ограничивать синтез гема и образование важных гемопротеинов. Например, снижение активности NO-синтазы может уменьшить производство NO и вызвать вазоспазм, что объясняет некоторые церебральные проявления ОПП. Однако регулирование синтеза гема и гемопротеинов в нервной ткани и кровеносных сосудах трудно изучать, и на данный момент убедительных доказательств не хватает.
- Нарушение синтеза гема в печени во время острых атак может привести к снижению активности триптофанпирролазы, которая может увеличить уровни триптофана в плазме и головном мозге, что приведёт к усилению синтеза серотонина и накоплению нейротоксических доз.
Диагноз острая порфирия верифицируется при помощи количественного определения порфиринов и их предшественников в моче. Для острых приступов характерно высокое содержание общих порфиринов и их предшественников: аминолевулината и порфобилиногена в моче. Кроме того, высокие показатели 2 вышеперечисленных предшественников гема часто остаются высокими в период латентного течения, что является отличительной чертой ОПП при сравнении с другими формами острых порфирий. По сочетанию характерных клинических признаков с увеличенным содержанием общих порфиринов и их предшественников в моче, кале и крови, а также со сниженной активностью ПБГД в эритроцитах удаётся установить правильный диагноз больным ОПП. Для асимптомных носителей дифференциальная биохимическая диагностика затруднительна. Измерение активности ПБГД в эритроцитах далеко не всегда даёт однозначный ответ на вопрос о наличии заболевания, поскольку диапазоны уровней активности фермента у таких пациентов перекрываются с нормальными значениями. В этом случае единственным способом уточнить диагноз является молекулярно-генетическое исследование для выявления мутации в гене ПБГД (рис. 4: схема дифдиагностики).
Рисунок 4.
Если диагноз установлен, для профилактики острых атак следует исключить приём потенциально опасных лекарств. У женщин атаки могут быть связаны с лютеиновой фазой менструального цикла. Для подавления овуляции им назначают аналоги гонадотропин-рилизинг гормона (ГРГ). Вследствие приёма аналогов ГРГ возможно снижение плотности костной ткани. Для предотвращения переломов назначаются бисфосфонаты. Кроме того, пациентам старше 50 лет рекомендуется длительный мониторинг функции печени, почек и уровня АД.
При атаке необходимо немедленно начать купирование тяжёлых симптомов, инфузионную терапию и коррекцию электролитных нарушений, мониторинг жизненно важных функций. По возможности устраняют провоцирующие факторы и начинают симптоматическую терапию:
- анальгетики — для купирования болевого синдрома;
- бензодиазепины короткого действия в низких дозах — для устранения психических нарушений;
- хлорпромазин — для купирования тошноты и рвоты;
- бета-блокаторы — для контроля гипертензии и тахикардии.
Относительно безопасными противосудорожными препаратами являются габапентин, вигабатрин и клоназепам. При тяжёлом течении показана внутривенная инфузия гемина, который по отрицательной обратной связи подавляет транскрипцию печеночной аминолевулинатсинтазы (ALAS1). Это снижает образование предшественников порфирина.
Наследственная копропорфирия наследуется по аутосомно-доминантному типу, часто протекает латентно из-за низкой пенетрантности. НКП является наименее распространённой среди острых порфирий. При этой болезни обнаруживается нарушение активности копропорфириногеноксидазы, митохондриального фермента, который превращает копропорфириноген III в протопорфириноген IX. Когда активность фермента нарушена, накапливается копропорфириноген III, который выделяется с фекалиями и мочой. НКП по клиническим проявлениям напоминает острую перемежающуюся порфирию.
НКП является острой печеночной порфирией с выраженными нейровисцеральными симптомами. Острые атаки часто связаны с использованием определённых препаратов, голоданием и менструацией. Атаки обычно начинаются со слабой боли в животе, которая медленно усиливается в течение нескольких часов или дней, часто сопровождается тошнотой и рвотой. Обычно боль плохо локализована, но в некоторых случаях имитирует острое воспаление жёлчного пузыря, аппендикса или другого внутрибрюшного органа. Иногда боль преобладает в спине или конечностях. Моторная невропатия может развиваться в течение нескольких дней или недель, если отсутствует лечение. Дыхательная недостаточность может быть следствием нарушения иннервации диафрагмы и респираторных мышц. Кожные проявления встречаются очень редко. Следует сказать, что у пациентов с любым типом острой порфирии, если они гетерозиготны по соответствующим мутантным генам, в почках и печени могут развиться хронические изменения, которые часто являются субклиническими. Одним из проявлений этих изменений в печени является гепатоцеллюлярная карцинома, которая возникает в основном после 60 лет [3,4].
Диагноз ставится после количественного анализа порфиринов как в моче, так и в фекалиях. В моче определяется трёхкратное увеличение порфобилиногена и копропорфирина. Молекулярно-генетические тесты используются для подтверждения диагноза. Дифференциальная диагностика должна включать другие печёночные порфирии, отравление свинцом, синдром Ротора (наследственный пигментынй гепатоз) и неспецифическая копропорфиринурию, в которых также имеется повышение уровня предшественников порфирина [2] (рис.4).
Острые приступы лечатся путём прекращения приёма любых лекарств, которые, как считается, их вызывают. Затем начинают симптоматическую и инфузионную терапию, как при ОПП.
Это заболевание называют «вариегатным» (пёстрым), потому что оно может вызывать тяжелые нейровисцеральные симптомы или фотодерматоз, идентичные тем, которые наблюдаются при поздней кожной порфирии. ВП в литературе также упоминается как смешанная или южноафриканская генетическая порфирия. ВП является аутосомно-доминантным генетическим расстройством из-за недостаточной активности митохондриального фермента протопорфириногеноксидазы (ррох). Распространённость ВП составляет 1:300 среди белого населения голландского происхождения в Южной Африке из-за эффекта основателя. Более низкая распространённость ВП была оценена в Финляндии (1,3:100000) и Европе в целом (0,3:100000) [2].
Клинические проявления чаще встречаются у женщин, чем у мужчин. Бывают острые приступы, идентичные тем, которые встречаются при ОПП. Характерными являются боли в животе, груди и конечностях, гипертония, тахикардия, беспокойство, судороги и нервно-мышечная слабость, которая может прогрессировать до квадриплегии и дыхательного паралича. Фототоксические проявления могут включать субэпидермальные везикулы, буллы, эрозии или язвы, которые медленно заживают.
Диагноз ВП устанавливается путём биохимического тестирования и подтверждается идентификацией гетерозиготной мутации в ррох (таб. 2). Тактика ведения пациентов с оcтрыми атаками такая же, как при ОПП. Следует избегать солнца, поскольку эффективное лечение кожных проявлений отсутствует. Важно различать ВП от поздней кожной порфирии, потому что кожные проявления у них очень похожи [4].
Таблица 2.
ПКП представляет собой печёночную порфирию, которая проявляется в виде фотодерматоза и не имеет никаких неврологических признаков. Следовательно, это заболевание отличается от острых печёночных порфирий, о которых говорилось выше. Поскольку ПКП является наиболее распространённой порфирией, в разных странах в диапазоне от 1:5000 до 1:70000, важно отличать её от других порфирий, которые имеют кожные проявления. Болезнь обычно манифестирует в среднем/пожилом возрасте, поэтому называется поздней. Она также известна под многими другими названиями, включая такие, как симптоматическая порфирия, идиосинкразическая порфирия, химическая порфирия или приобретённая печеночная порфирия [4,5].
Это единственная порфирия, которая может развиться в отсутствие наследуемой мутации поражённого фермента. Только 20% пациентов с этой болезнью имеют гетерозиготную мутацию urod (уропорфириногендекарбоксилазы), которая снижает активность urod во всех тканях до половины от нормы. Эти мутации повышают восприимчивость к развитию ПКП, но необходимо присутствие дополнительных факторов. Всё это приводит к тому, что активность фермента снижается на 20% от нормального уровня и болезнь манифестирует. Таким образом, ПКП является гетерогенным расстройством, в котором взаимодействуют экологические, инфекционные и генетические факторы, вызывающие существенный дефицит активности фермента в печени. Заболевание имеет 3 формы в зависимости от наличия мутаций в гене urod [3,5]:
- спорадическая форма. Самая частая форма поздней кожной порфирии без мутации urod;
- семейная форма. Пациенты гетерозиготны по мутациям гена urod, наблюдается частичное снижение активности фермента, передаётся аутосомно-доминантно. Сейчас некоторые авторы её называют гепатоэритропоэтической порфирией. Она манифестирует в детском возрасте не только тяжёлыми кожными проявлениями, но и гемолитической анемией, в какой-то степени напоминая ВЭП (рис. 5);
- приобретённая форма. Видимо, существует генетическая предрасположенность, которая приводит к снижению активности фермента в гепатоцитах. Считается, что в развитии этой формы задействованы ещё не идентифицированные гены, отличные от urod [3,4].
Рисунок 5.
В патогенезе ПКП важную роль играет образование ингибитора уропорфириногендекарбоксилазы, который образуется после окисления уропорфириногена или гидроксиметилбилана. Этот процесс требует присутствия высокого уровня железа, активности CYР-4501А2 и эстрогенов. Но точная структура и характер ингибитора остаются неизвестными. Перегрузка железом и длительный приём эстрогенов являются дополнительными факторами, вызывающими манифестацию латентной ПКП по неизвестным механизмам. Об этом свидетельствуют данные о том, что больше половины пациентов имеют гемосидероз и повышенный уровень плазменного ферритина. Кроме того, удаление железа из организма возвращает активность фермента в нормальный уровень в спорадической форме болезни [4].
Основными факторами риска ПКП являются чрезмерное потребление алкоголя, курение, перегрузка железом, вирус гепатита С, ВИЧ и приём эстрогенов (оральные контрацептивы, лечение рака простаты). Один или несколько из этих факторов могут быть обнаружены у более 80% пациентов. Генетическая предрасположенность является важным фактором, но она сама по себе недостаточна для развития ПКП, необходимы и другие факторы [5].
Существует несколько предложенных механизмов, по которым алкоголь приводит к снижению активности уропорфириногендекарбоксилазы. Алкоголь увеличивает абсорбцию железа, что приводит к накоплению железа в печени, активирует печёночный ALAS-1, образование свободных радикалов и является гепатотоксичным. Алкоголь также ингибирует другие ферменты в пути синтеза гема. Алкоголь, наряду с другими вышеперечисленными факторами, вызывает дефицит фермента, что приводит к накоплению порфиринов в печени [5].
Было показано, что вирусный гепатит С и ВИЧ-инфекция связаны с ПКП. Роль гепатита С не совсем понятна. Возможная гипотеза заключается в том, что он вызывает клинические проявления болезни у генетически предрасположенных лиц. Что же касается ВИЧ-инфекции, то она каким-то образом влияет на биосинтез гема и нарушает функционирование системы цитохрома Р-450.
Уропорфириногендекарбоксилаза катализирует ступенчатое декарбоксилирование уропорфириногена в копропорфириноген. Снижение активности фермента приводит к увеличению продуцирования уропорфиринов. Эти соединения автоматически окисляются до порфиринов, которые накапливаются в печени в больших количествах и через кровь переносятся на кожу, где они действуют как фотосенсибилизаторы. Они взаимодействуют со светом длиной волны около 400 нм; активируясь, генерируют активные формы кислорода, которые вызывают повреждение кожи — буллы, милии (рис. 6). Хроническое повреждение кожи может привести к образованию рубцов и изменению кожной пигментации. Другие кожные проявления могут включать гелиотропную гиперемию периорбитальных областей, хлоракне, гипертрихоз, алопецию и онихолизис [5].
Рисунок 6.
У пациентов моча имеет розово-красный цвет. Диагноз ставится путём нахождения существенного повышения количества порфиринов в моче или плазме с преобладанием карбоксилированных порфиринов (уропорфирин и гепта-, гекса- и пентакарбоксипорфирины). Копропорфирины также увеличиваются. Уровни дельта-аминолевулиновой кислоты и порфобилиногена в моче нормальны, либо слегка повышены. Как правило, лица с ПКП имеют слегка повышенные уровни аминотрансфераз в сыворотке и уровень гамма-глутамилтранспептидазы тоже повышен. Неспецифические результаты обычных лабораторных тестов дают понять, что для диагностики ПКП необходимы специальные исследования. Высокая чувствительность у флуометрических методов. Последние помогают отличать ПКП от других печёночных порфирий (наследственная копропорфирия, вариегатная порфирия), которые также имеют кожные проявления [4,5].
Как и при других типах кожной порфирии, избегание воздействия солнца является наиболее эффективным способом предотвращения кожных проявлений. Терапия направлена на патогенетический механизм ПКП и включает снижение уровня железа, прием противомалярийных средств, которые удаляют порфирины из печени и других тканей, и флеботомию. Пациентам в первую очередь рекомендуют прекращение курения, употребления алкоголя и приёма эстрогенов. Кроме того, рекомендуется соблюдение диеты для предотвращения компенсаторного увеличения абсорбции железа в ЖКТ. Флеботомия является достаточно безопасным и эффективным методом лечения. Она заключается в том, что еженедельно или ещё реже забирают 400‒500 мл крови до тех пор, пока уровень сывороточного ферритина не станет <25 мкг/л. Клиническая ремиссия обычно проявляется после удаления 2‒4 л крови. Кожные проявления постепенно улучшаются с разрешением булл за 2‒3 месяца, а полная биохимическая ремиссия наблюдается после 13 месяцев. При флеботомии необходимо соблюдать осторожность, чтобы предотвратить снижение уровня гемоглобина ниже 110 г/л. Предсуществующая анемия, тяжёлая сывороточная белковая недостаточность (например, из-за цирроза и кровотечений) являются относительными противопоказаниями к флеботомии [5].
Если флеботомия противопоказана, низкая доза хлорохина (125 мг через день) является следующим методом выбора. Хлорохин связывает карбоксипорфирины, которые накапливаются в лизосомах, и, образуя с ними водорастворимые комплексы, он приводит к увеличению их экскреции мочой. Однако он гепатотоксичен. Подкожное введение хелатора железа — дефероксамина — также является альтернативой флеботомии. Хотя это не так дёшево, как терапевтическая флеботомия, дефероксамин способен удалять токсическое железо, вовлечённое в патогенез болезни. Кроме того, он может уменьшить компенсаторное увеличение поглощения железа в кишечнике, часто наблюдаемое при ремиссии, вызванное большей степенью снижения железа, достигаемой флеботомиями. Оральные хелаторы железа (деферазирокс, деферипрон) доступны в некоторых странах. Они также могут быть эффективными и относительно безопасными для лечения ПКП [5].
Источники:
- Пустовойт Я. С. Федеральные клинические рекомендации по диагностике и лечению острых порфирий. – 2013.
- DiMauro S. et al. Gene Reviews //University of Washington: Seattle, WA, USA. – 2010.
- Kaushansky K., Lichtman M. A. Williams Hematology. – 2021. – C. 2528.
- Ramanujam V. M. S., Anderson K. E. Porphyria diagnostics—part 1: a brief overview of the porphyrias //Current protocols in human genetics. – 2015. – С. 17.20. 1-17.20. 26.
- Ryan Caballes F., Sendi H., Bonkovsky H. L. Hepatitis C, porphyria cutanea tarda and liver iron: an update //Liver International. – 2012. – Т. 32. – №. 6. – С. 880-893.
Анализы и диагностика
В целях диагностики используются специальные методы лабораторного определения порфиринов/их предшественников в кале и моче, и в крови — для оценки активности ферментов.
Так, для болезни Гюнтера в клинико-биохимических анализах крови характерны признаки гемолиза — повышение сывороточного железа/непрямого билирубина, пойкилоцитоз, ретикулоцитоз, сфероцитоз, анизоцитоз, увеличение печеночных трансаминаз. Для пациентов с острыми порфириями характерно снижение уровня натрия/глюкозы, при поздней кожной порфирии— увеличение ферритина/сывороточного железа, положительные маркеры на вирус гепатита С.
При постановке диагноза важен семейный анамнез: наличие у близких родственников заболевания, а также обстоятельства начала заболевания (беременность, менструации, инсоляция, прием алкоголя/лекарств, голодание, инфекции и др.). Для подтверждения диагноза определяются уровни различных ферментов цикла биосинтеза гема в плазме, эритроцитах, лимфоцитах.
Из инструментальных методов могут назначаться КТ/УЗИ органов брюшной полости, КТ/рентгенография грудной клетки, ЭКГ, ЭЭГ головного мозга и др.
Клинический случай
Пациент Т
., 20 лет, 23.12.17 во время тренировки по спортивным единоборствам получил удар в живот, после чего отмечал эпизод тошноты. Через 4 дня появились выраженная боль в эпигастральной области, тошнота, рвота желчью. Лечение не получал, в течение 2 дней симптоматика регрессировала. 05.01.18 вновь почувствовал боль в эпигастральной области, сопровождавшуюся рвотой и жидким стулом, температура тела при этом не повышалась. Через 2 сут госпитализирован в районную больницу по месту жительства с диагнозом «острый панкреатит». Перенесенные ранее заболевания и обращения за медицинской помощью отрицал. Назначена антисекреторная терапия, рекомендован голод. За 2 нед потерял массу тела на 13 кг, появились слабость и боли в конечностях. 16.01.18 г. пациент переведен в отделение реанимации и интенсивной терапии (ОРИТ) областной больницы. При обследовании обнаружены электролитные нарушения (уровень натрия — 128 ммоль/л, калия — 2,8 ммоль/л), амилаземия (уровень амилазы — 223 МЕ/л), стеноз устья чревного ствола (80% по данным компьютерной томографической ангиографии). Консервативная терапия к улучшению состояния пациента не привела, в связи с чем 28.01.18 он госпитализирован в Федеральный научно-клинический центр специализированных видов медицинской помощи и медицинских технологий с диагнозом «Стеноз чревного ствола. Синдром хронической абдоминальной ишемии. Кахексия. Электролитные нарушения» для решения вопроса об оперативном лечении. При поступлении: состояние пациента тяжелое. Кожные покровы обычной окраски, нормальной влажности. Температура тела 37,7 °С. Дыхание самостоятельное, везикулярное, с частотой 18 в 1 минуту, насыщение гемоглобина крови кислородом 98%. Артериальное давление — 155/110 мм рт.ст., частота сердечных сокращений — 115 уд/мин. По данным электрокардиографии — синусовая тахикардия. Живот не вздут, при пальпации мягкий, безболезненный, симптомов раздражения брюшины не было. Энтеральное питание усваивал, отмечались снижение аппетита, запоры в течение 1 нед. Мочеиспускание не контролировал, цвет мочи соломенно-желтый. По поводу стеноза чревного ствола больной консультирован сосудистым хирургом, от хирургической тактики решено воздержаться, рекомендовано консервативное лечение. При осмотре неврологом: сознание ясное, по шкале комы Глазго (ШКГ) — 15 баллов. Ориентация в месте, времени, собственной личности полная. Менингеальные симптомы отрицательные. Зрачки OD = OS, реакция на свет в норме. Движения глазных яблок в полном объеме. Лицо симметричное, язык по средней линии. Дизартрии, дисфонии, дисфагии не было. Мышечный тонус снижен. Тетрапарез по шкале оценки мышечной силы: в руках до 2 баллов в проксимальных и до 4 баллов в дистальных отделах, в ногах — до 3 баллов в проксимальных и дистальных отделах. Сухожильные рефлексы не вызывались. Нарушений чувствительности не было. Имелись нарушения тазовых функций по типу недержания мочи, запоров. Неврологом рекомендовано дообследование (электронейромиография (ЭНМГ), магнитно-резонансная томография (МРТ) головного мозга, шейного и грудного отделов позвоночника, исследование ликвора) для дифференциальной диагностики и уточнения генеза полинейропатии. При лабораторном обследовании обнаружены лейкоцитоз, анемия легкой степени и тромбоцитопения (лейкоциты 13,3·109/л, гемоглобин 12,3 г/дл, тромбоциты 114·109/л соответственно), повышение уровня панкреатической амилазы (77 Ед/л), гипоальбуминемия (30 г/л), электролитные нарушения (натрий 124 ммоль/л, калий 3,5 ммоль/л), повышение уровней воспалительных маркеров (С-реактивный белок 47,6 мг/л, прокальцитонин 7,4 нг/мл), удлинение активированного частичного тромбопластинового времени (40 с), снижение сывороточной концентрации железа (2,2 мкмоль/л) и фолиевой кислоты (1,9 нг/мл). При компьютерной томографии (КТ) органов грудной полости выявлены единичные очаги в нижних долях легких.
Начаты коррекция водно-электролитных нарушений (инфузии растворов натрия хлорида 0,9% и калия хлорида 4%) и анемии (фолиевая кислота, сульфат железа), эмпирическая антимикробная терапия (левофлоксацин), смешанное питание, профилактика стресс-язв (омепразол), профилактика тромботических осложнений (эноксапарин натрия), мероприятия по реабилитации в ОРИТ, включая механотерапию в прикроватных тренажерах, лечебную физкультуру (ЛФК), высаживание в прикроватное кресло.
В рамках дообследования при ЭНМГ обнаружена аксонально-демиелинизирующая (первично аксональная) моторно-сенсорная полинейропатия. При МРТ данных за стенозирование шейного и грудного отделов позвоночного канала, наличие объемных образований, очаговое поражение головного мозга не получено. Анализ ликвора от 30.01.18: бесцветный, прозрачный, цитоз 2 кл/мкл, глюкоза 3,9 ммоль/л, белок 0,2 г/л; вирусологические данные (вирус Эпштейна—Барр, цитомегаловирус, вирус простого герпеса 1-го и 2-го типов) — отрицательные. При бактериологическом исследовании крови, дистального конца удаленного венозного катетера и мокроты обнаружен рост золотистого стафилококка, в связи с чем с 30.01.18 левофлоксацин заменен на линезолид.
Учитывая результаты дообследования, состояние пациента расценили как проявление синдрома Гийена—Барре и кахексии с алиментарной недостаточностью вследствие перенесенного панкреатита, осложнившихся развитием нозокомиальной пневмонии, а также катетерассоциированной инфекции кровотока и тромбоза яремной и подключичной вен. 31.01.18 начат 5-дневный курс специфической терапии синдрома Гийена—Барре иммуноглобулином G — препаратом Октагам («Octapharma Pharmaceutica Production GmbH», Австрия) в дозе 0,4 мг на 1 кг массы тела в сутки.
Несмотря на проведенное лечение и ежедневные реабилитационные мероприятия, неврологическая симптоматика сохранялась на прежнем уровне. 05.02.18 усилились боли в конечностях. Учитывая длительный анамнез иммобилизации в ОРИТ, симптоматику расценили как проявления ПИТС, и к терапии добавлены флувоксамин и карбамазепин. Кроме того, сохранялись электролитные нарушения, и с целью исключения надпочечниковой недостаточности пациент дополнительно обследован: суточная экскреция натрия с мочой составила 87 ммоль/л, сывороточный уровень кортизола 19,7 мкг/дл. Учитывая полученные данные, к терапии добавили гидрокортизон. В связи с неэффективностью монотерапии линезолидом (повторный рост золотистого стафилококка в крови 05.02.18, сохраняющаяся высокая температура тела), антимикробная терапия усилена цефазолином. При повторном дуплексном сканировании отмечены сохраняющиеся тромбозы подключичной и внутренней яремной вен, что, вероятно, и служило причиной стойкой бактериемии, несмотря на проводимую антимикробную терапию.
Для исключения инфекционного эндокардита выполнена чреспищеводная эхокардиография, при которой данных за наличие вегетаций на клапанном аппарате и в полостях сердца не получено. Во время эзофагогастродуоденоскопии обнаружен грибковый эзофагит, позже подтвержденный результатами микроскопии.
После начала терапии карбамазепином и флуконазолом 08.02.18 состояние пациента с отрицательной динамикой в виде снижения уровня сознания до оглушения (14 баллов по ШКГ), появления спонтанного нистагма, тошноты. При повторной МРТ головного мозга с целью исключения понтинного миелинолиза изменений не выявлено. На следующие сутки восстановилось ясное сознание, однако пациент стал эмоционально лабилен, начал жаловаться на нарастающие ноющие боли в конечностях и сердце, при этом изменений на электрокардиограмме не было.
По совокупности всех клинико-лабораторных и инструментальных данных у пациента заподозрена острая порфирия, что потребовало специального лабораторного подтверждения. 09.02.18 в анализе мочи обнаружен порфобилиноген (ПБГ) 200,6 мг/л, δ-аминолевулиновая кислота (АЛК) 145,6 мг/л. Пациент переведен в отделение реанимации Национального медицинского исследовательского центра гематологии, где находился в течение 25 сут. В рамках патогенетической терапии острой порфирии проводилась постоянная инфузия 20% раствора глюкозы в объеме 1000 мл/сут, проведен курс аргинатом гема (препарат Нормосанг, «Orphan Europe S.a.r.L.», Франция) 125 мг/сут. В динамике в результате проводимой терапии отмечено снижение концентрации ПБГ с 200 до 31 мг/л, АЛК с 146 до 23 мг/л. Пациенту продолжена антибактериальная терапия стафилококковой инфекции (очаговая двусторонняя пневмония, инфекция кровотока). Диагностирован пиелонефрит, вызванный Pseudomonas aeruginosa.
Проводилась терапия тромбозов вен нижних конечностей и яремной вены справа. В связи с сохраняющейся алиментарной недостаточностью пациент получал смешанное питание. Течение заболевания в отделении реанимации осложнилось развитием симптоматического психоза, в связи с чем проводилась терапия галоперидолом с положительным эффектом. По данным лабораторных исследований выявлены признаки фолиеводефицитной и железодефицитной анемии, проводилась заместительная терапия фолиевой кислотой. На фоне коррекции гипонатриемии удалось стабилизировать уровень натрия в пределах 132 ммоль/л (минимальный уровень достигал 120 ммоль/л). Ежедневно проводились занятия кинезиотерапией и ЛФК. 04.04.18 пациент был выписан. Его состояние расценивалось как средней степени тяжести и обусловлено течением основного заболевания, сохраняющимся вялым парезом конечностей, алиментарной недостаточностью, анемией сочетанного генеза, тромботическими осложнениями.
Профилактика
Специфическая профилактика отсутствует. С целью минимизации приступов рекомендуется:
- Избегать/минимизировать воздействие порфириногенных факторов (режимный образ жизни, избегать стрессов, приема потенциально опасных лекарств, употребления алкогольсодержащих напитков, голодания/строгих диет, пребывания на солнце).
- Пациенты старше 50 лет должны проходить дважды в год обследование для оценки состояния печени.
- Рациональное трудоустройство.
- Проведение профилактического лечения/диспансеризация больных.
- При наличии в семье больного порфирией необходимо проводить ДНК-диагностику (выявление генетических мутаций) и определять активность различных ферментов цикла синтеза гема.
Последствия и осложнения
Поскольку порфирии за исключением поздней кожной порфирии являются наследственно обусловленными их излечение полностью невозможно. При наличии у пациента 2-3 и более приступов в год может длительно сохраниться неврологический дефицит в виде вялых периферических парезов. В случаях тяжелого течения возможны осложнения в виде бульбарного синдрома, гипонатриемии, паралича дыхательной мускулатуры с высокой угрозой летального исхода. При порфириях с поражением кожных покровов — риск развития склеродермоидных процессов.
Общая информация
Порфириновая болезнь диагностируется сравнительно редко: российские врачи выявляют не более 12 случаев на 100 тысяч человек. Различные формы патологии получают распространение в отдельных регионах Земли. Так, признаки поздней кожной порфирии часто выявляется у жителей Южной Африки (1 случай на 800 человек). Острый перемежающийся тип заболевания характерен для жителей Северной Европы (1 случай на 1000 человек). Мужчины и женщины одинаково часто страдают от различных форм синтеза гема.
Список источников
- Кузнецова Н.П., Панков Б.С., Чубарова А.С. и др. Порфирии. М., 1981. с. 66–14.
- Пустовойт Я.С., Пивник А.В., Карпова И.В. Клиника, диагностика и лечение порфирий // Пособие для врачей. Москва, 2003 г.
- Воробьёв А, Кравченко С, Кременецкая А, Карпова И, Пустовойт Я. Острая перемежающаяся порфирия: проблемы диагностики и лечения // Врач. 2003 г. №2, стр. 8-13.
- Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. — 2021.
- Руководство по гематологии в 3 томах/ под ред. Воробьева А.И. – 2005 – Т.3.