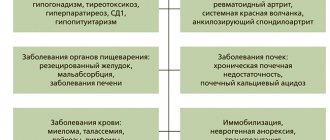
Due to a deficiency in the production of this hormone (due to decreased calcium absorption in the intestine), the balance between sodium and potassium, calcium and magnesium ions is disrupted, neuromuscular excitability increases, paroxysmal involuntary (tonic) convulsions develop, and the functioning of the gastrointestinal tract changes.
The disease in various forms occurs in 2-3 people out of 1000, regardless of gender and age, and consists of a violation of phosphorus-calcium metabolism. As an independent disease, hypoparathyroidism of various origins occurs in 0.2–0.3% of the population.
Possible reasons
Insufficient secretion of specific parathyroid hormone by the endocrine glands (parathyroid), or insensitivity of the cells of individual organs to this hormone, leads to disturbances in the metabolism of calcium and phosphorus.
Clinical causes of pathology also arise under the influence of provoking factors that have any effect on the parathyroid glands:
operations performed on the cervical area, especially on the thyroid gland, with complications in the form of iatrogenic damage;
- neck trauma with bleeding;
- inflammatory processes;
- metastasis in endocrine organs;
- DiGeorge syndrome;
- ionizing radiation;
- adrenal insufficiency;
- complications after treatment of primary hypothyroidism;
- a number of autoimmune diseases;
- systemic diseases.
Metabolic processes between calcium and phosphorus are supported by the complex regulation of calcitonin, parathyroid hormone and vitamin D. With hormonal deficiency, an increase in phosphate in the blood occurs with a concomitant decrease in calcium levels.
An imbalance of minerals leads to neuromuscular excitability and convulsive attacks, and a violation of calcium metabolism leads to the deposition of salts on the vascular walls and tissues of internal organs.
Hypoparathyroidism in childhood and adolescence
Part 1
Hypoparathyroidism (hypoPT) is an endocrine disorder characterized by insufficient synthesis, secretion, or peripheral action of parathyroid hormone (PTH). The consequence of hypoPT is a disturbance of phosphorus-calcium metabolism. Hypocalcemia is the main symptom of this disease, but its causes can be different.
Calcium (Ca) plays an important role in the functioning of any cell and in the metabolic processes of the body as a whole. It is involved in muscle contraction, is an intermediary in the transmission of signals between cells, and changes the permeability of cell membranes.
Bone tissue is considered the main Ca storage in the body (about 99%). Approximately 1% of it is in the form of labile phosphate salts, which can easily dissolve and change the concentration of Ca in the blood plasma.
The fraction of free (ionized) calcium (Ca2+) is biologically active (approximately 50%), the rest of Ca is associated with blood plasma proteins, citrates, phosphates, which affect bone density. An alkaline environment increases the bond of Ca with albumin, reducing the pool of the ionizing fraction.
Phosphorus in the human body is contained in crystalline form in bone tissue (85%), in the intercellular fluid in the form of inorganic phosphate and in soft tissues in the form of phosphorus esters.
Together with calcium, phosphorus forms the basis of bone tissue. Phosphates are involved in energy transfer in the form of macroergic bonds (TP, ADP, creatine phosphate, etc.), and are part of the buffer systems of plasma and tissue fluid. Phosphoric acid affects the processes of glycolysis, gluconeogenesis, and fat metabolism.
In addition to Ca ions, the coupled processes of excitation and contraction also require cyclic nucleotide adenosine 3–5 monophosphate (cAMP). It is a secondary messenger in the reduction process. Ca and cAMP are bound to each other. Ca regulates the rate of synthesis and breakdown of cAMP, at the same time the latter controls the entry of Ca ions into the cell and thus is a regulator of the cycle of muscle contraction and relaxation [1].
Anatomy and physiology of the parathyroid glands
The parathyroid glands (PTG) are formed in the embryo at the 5th week of intrauterine development from 3–4 pairs of gill pouches of the pharyngeal intestine. Subsequently, these protrusions are laced to form 4 separate parathyroid glands. Additional parathyroid glands are found in the tissue of the thyroid and thymus glands, in the anterior and posterior mediastinum, in the pericardium, behind the esophagus, in the area of the bifurcation of the common carotid artery.
Each pancreas is surrounded by a connective tissue capsule. The parenchyma of the gland is represented by trabeculae and clusters of endocrine cells - parathyrocytes - between them.
PTH is synthesized in the main acidophilic cells. The gene encoding the synthesis of PTH is localized on chromosome 11 (11p15). Vitamin D (1,25(OH)2D3), which, like calcium, can act independently of each other, takes part in the regulation of PTH gene expression.
First, a preprohormone is synthesized, containing 115 amino acid residues. Next, a signal peptide containing 25 amino acid residues is cleaved from it, and a prohormone is formed, which moves to the Golgi apparatus. It is converted into a mature hormone, stored in secretory granules.
PTH circulates in the blood in three main forms: intact parathyroid hormone with a molecular weight of 9500, a biologically active carboxyl fragment with a molecular weight of 7000–7500, and a biologically active fragment with a molecular weight of 4000. The formation of fragments occurs in the liver and kidneys. The intact hormone subsequently breaks down into short units that bind to receptors on target cells [1].
Regulation of PTH secretion occurs with the help of calcium-sensitive receptors (CaSR) on the surface of parathyroid cells and is pulsating in nature: for a short time through ionized Ca, and with long-term stimulation together with vitamin D. PTH secretion is maximum at low Ca2+ levels, an increase in the latter suppresses her [2]. The biological half-life of PTH in the body is about 18 minutes. PTH is completely inactivated within a few hours in Kupffer cells of the liver and kidneys. Chromographin is also produced in the pancreas.
At birth, the PTG is well developed and functionally active, but with sufficient transplantar calcium transport, PTH secretion is reduced. An increase in PTH concentrations in the blood serum of a newborn during the early adaptation period occurs simultaneously with a decrease in Ca concentrations. Normal or elevated PTH levels in healthy newborns are established within 2–3 days [3].
The target organs for PTH are bones and kidneys. They contain calcium-sensitive receptors, acting on which PTH activates adenylate cyclase. It affects ATP, converting it into cyclic adenosine monophosphate (cAMP), which in turn mobilizes calcium ions from intracellular stores.
PTH controls the concentration of divalent cations circulating in the blood, mainly Mg2+ and Ca2+, as well as the content of cAMP in the cells of the pancreas.
Magnesium is involved, in particular, in the activation of mitochondrial ATP synthetase, regulates the activity of some ion channels and, having the ability to form stable three-component complexes with nucleotides, participates in the processes of nucleic acid synthesis, transcription and translation [4].
The introduction of large amounts of magnesium into the body significantly reduces the secretion of PTH, which is used clinically for hyperparathyroidism.
The main function of PTH is to maintain a constant level of ionized Ca in the blood, which it performs by influencing bone tissue, kidneys and indirectly through vitamin D in the intestines.
In bones, PTH activates resorptive processes. Bone tissue contains three types of cells: osteocytes, osteoblasts (young cells that create bone tissue) and osteoclasts (destroy bone tissue). In bone tissue, PTH receptors are located only on osteocytes and osteoblasts. When PTH binds to osteoblasts, they begin to intensively synthesize insulin-like growth factor 1 (IGF1) and various cytokines. These substances affect osteoclasts, stimulating their metabolic activity.
Osteoclasts synthesize proteinases, such as alkaline phosphatase and collagenase, which influence the breakdown of bone matrix. As a result, Ca and phosphates are mobilized into the extracellular fluid. This effect is associated with an increase in calcium levels in the blood. The effect of PTH on bone tissue occurs only in the presence of vitamin D3 (cholecalciferol).
In the kidneys, PTH, like vitamin D, enhances the reabsorption of Ca in the distal tubules, thereby reducing its excretion in urine and increasing its level in the blood. The mechanism of action of PTH on phosphate in the renal tubules is directly opposite to the effect of vitamin D. The former suppresses the reabsorption of phosphate in the nephrons, on the contrary, vitamin D enhances its reabsorption [3]. The phosphaturic effect of parathyroid hormone prevents the deposition of calcium phosphate in soft tissues.
Vitamin D acts in the intestine to increase the production of calcium-binding protein, which is necessary for the transport of exogenous Ca across the cell membrane of the intestinal mucosa.
The synthesis of the active form of vitamin D3 (1α,25(OH)2D3 - D-hormone - calcitriol) occurs with the participation of the enzyme 1a-hydroxylase in the proximal renal tubules under the control of PTH. The synthesis of this key enzyme also requires sex hormones, thyroid hormones, somatotropic hormones, prolactin, and calcitonin.
The main physiological function of vitamin D (its active metabolites) in the body is to regulate and maintain phosphorus-calcium homeostasis at the required level and ensure bone mineralization. In bone, 1α,25(OH)2D3 binds to receptors on osteoblasts, increasing the expression of the nuclear factor activator RANKL. This accelerates the maturation of preosteoclasts and their transformation into mature osteoclasts [5].
In bone tissue, calciferol mobilizes calcium and uses it for the mineralization processes of newly formed bone. Along with this, it affects the synthesis of collagen, which is involved in the formation of the bone tissue matrix. The mechanism of calcium absorption in the intestine is associated with the synthesis of calcium-binding protein (CaBP) by enterocytes, one molecule of which transports 4 calcium atoms. Normally, a person consumes about 1 gram of Ca per day. From 25% to 50% of calcium is absorbed with the participation of calciferol.
So, calcium enters the blood plasma from the intestines, absorbed from water, food or through bone resorption. Serum calcium levels are important for a large number of physiological processes, since even small deviations affect various cellular functions. Serum calcium levels are usually maintained within a very narrow range.
Pathogenesis
An absolute or relative lack of PTH leads to a decrease in the activity of osteoclasts, a decrease in bone resorption and, accordingly, the flow of calcium from the bone into the blood. In the kidneys, due to a lack of PTH, the synthesis of active vitamin D decreases, the reabsorption of phosphates increases, and the absorption of Ca decreases, resulting in the development of hyperphosphatemia and hypocalcemia.
Negative calcium and positive phosphorus balance disrupt electrolyte balance, changing the calcium/phosphorus and sodium/potassium ratios. This leads to a universal disruption of the permeability of cell membranes, in particular in nerve cells, and to a change in polarization processes in the area of synapses.
The resulting increase in neuromuscular excitability and general autonomic reactivity leads to increased convulsive readiness and tetanic crises. In the genesis of tetany, a significant role also belongs to disturbances in magnesium metabolism and the development of hypomagnesemia, which reduce PTH synthesis. This facilitates the penetration of sodium ions into the cell and the exit of potassium ions from the cell, which also helps to increase neuromuscular excitability. The resulting shift in the acid-base state toward alkalosis also has the same effect [6].
Nosological forms
Newborn tetany
Neonatal tetany is relatively common. Accompanied by hypocalcemia and often hypophosphatemia. It is possible that the etiological factor in the pathogenesis of neonatal hypocalcemia is target organ resistance to parathyroid hormone and transient hypoPT. In the blood of healthy newborns, the concentration of parathyroid hormone is either very low or not determined at all, and only on the 4th day after birth does a parallel increase in the blood content of PTH, bound and ionized calcium begin. During the neonatal period, the reserves of the pancreas in children are reduced. However, a decrease in calcium concentration in the blood is not always accompanied by an increase in PTG function, which indicates the participation of additional unknown factors in the genesis of neonatal hypocalcemia.
In newborns, hypocalcemia can manifest itself as poor weight gain and excessive regurgitation, generalized seizures, and acute cardiovascular failure [7].
Familial isolated hypoparathyroidism
Congenital isolated hypoparathyroidism is most often caused by hereditary disorders of the synthesis of parathyroid hormone. There are two types of the disease: infantile X-linked hypoparathyroidism (the gene is mapped to Xq26-q27) and familial hypoparathyroidism with an autosomal dominant type of inheritance (the gene is mapped to the short arm of chromosome 11) [8]. Sex-dependent forms of the disease have a favorable prognosis. Children in this group have hypoplasia or ectopia of the parathyroid gland, hypoplasia or absence of the thymus gland.
In children with congenital Pthyroid deficiency, the calcium content in the blood is reduced, the growth of bones, teeth and hair is impaired, and prolonged contractions of muscle groups (forearms, chest, pharynx, etc.) are observed.
Familial hypercalciuric hypocalcemia
Familial hypercalciuric hypocalcemia - This autosomal dominant hypocalcemia is caused by an activating mutation in the 3q13.3-q21 gene encoding the calcium-sensing receptor. Due to the activating mutation, CaSR sensitivity is high, which by feedback type leads to suppression of PTH secretion and a state of hypocalcemia. However, the thyroid gland in these patients functions normally, although the disease proceeds with the clinical picture of hypoPT.
Particular attention should be paid to patients with this form of the disease as they have abnormally high renal calcium excretion in response to treatment with calcium supplements, resulting in a high risk of developing renal calcification and renal failure [9].
Idiopathic or autoimmune hypoPT
Idiopathic, or autoimmune, hypoPT can be either isolated or part of multiple autoimmune endocrinopathy.
When isolated, autoantibodies are directed to calcium receptors on the membranes of PTG cells, namely to the associated censor, the G120 kDa protein, which detects the most minimal decrease in calcium levels in the blood serum. Since antibodies are associated with the G120 Kda protein, the latter does not stimulate PTH secretion to the required extent, its level in the blood serum is low, resulting in hypocalcemia [10]. Idiopathic autoimmune hypoPT syndrome is one of the rare forms of endocrinopathies, but cases of its development in childhood can occur.
Autoimmune polyglandular syndrome type 1
Autoimmune polyglandular syndrome type 1 (APS-1) is a monogenic disease with an autosomal recessive mode of inheritance; sporadic cases are less common. The syndrome is based on a mutation in the structure of the autoimmune regulator gene (AIRE).
The gene is located on chromosome 21q22.3, consists of 14 exons and is predominantly expressed in the thymus. Currently, more than 50 mutations of this gene have been identified. The most common are R257X, 109del13, R139X. These mutations affect regions of the gene that are responsible for the formation of DNA-binding domains [11].
There is no association of APS-1 with HLA halotypes. Peak manifestation occurs at 12 years of age. Characteristic signs of APS-1 are mucocutaneous candidiasis, primary chronic adrenal insufficiency and hypoPT.
The combination of symptoms, the timing of manifestation and the degree of their severity in APS-1 in children vary significantly. In most cases, the onset of the disease is accompanied by the development of mucocutaneous candidiasis in the first 10 years of life, often immediately after the birth of a child. In this case, damage to the mucous membranes of the oral cavity, genitals, skin, and nails is noted. Less commonly, the gastrointestinal tract, respiratory and urinary tract are involved in the process [8].
Against the background of candidiasis, 84% of patients are diagnosed with hypoPT, which usually develops in the first decade of a child’s life. The clinical manifestations of hypoPT are varied: from characteristic paresthesias and muscle cramps to the development of seizures similar to epilepsy. Primary hypocortisolism occurs in a latent form for a long time and manifests itself in most patients during the second decade of life as acute adrenal insufficiency.
Clinical phenotypes in this syndrome are heterogeneous and are not limited to these symptoms. The classic triad of the disease may be accompanied by primary hypogonadism, primary hypothyroidism against the background of autoimmune thyroiditis, and type 1 diabetes mellitus [12].
Among non-endocrine diseases in APS-1, alopecia, vitiligo, pernicious anemia, malabsorption syndrome, autoimmune hepatitis, abnormalities of tooth enamel, ectodermal dysplasia, isolated IgA deficiency, bronchial asthma, glomerulonephritis are detected. The appearance of the above clinical components of this syndrome can last for many years, which makes diagnosis difficult. Early diagnosis of APS-1 is possible in cases of long-term candidiasis and monoendocrine pathology when studying the AIRE gene [10].
Yartrogenic hypoparathyroidism
Yartrogenic hypoPT is the most common cause of post-surgical hypoPT (2–10%), which develops due to damage to the pancreas during neck surgery on the first day. According to Garrhyetal, hypoPT was associated with total thyroidectomy in 38% of cases, partial thyroidectomy in 9%, parathyroidectomy in 21%, and other neck surgeries in 5%. The risk of developing hypoPT after neck surgery largely depends on the experience of the surgeon [13]. Most patients developed transient hypoPT. Their PTG function was completely restored after 6 months. Chronic hypoPT developed in 15–25% [14].
Infiltrative-dystrophic hypoPT
Infiltrative-dystrophic hypoPT is associated with the deposition of various substances in the gland tissues in systemic diseases.
Hemochromatosis is a hereditary disease characterized by impaired iron metabolism in the body, impaired regulation of iron absorption in the intestine, increased iron content in the blood serum and accumulation in tissues and organs.
Thalassemia is a heterogeneous group of disorders resulting from decreased or absent production of normal globin chains. As a result, there is an increased breakdown of hemoglobin and excessive formation of the hemosiderin pigment and its deposition in the tissues of the gland.
Wilson–Konovalov disease is a hereditary disease transmitted in an autosomal recessive manner. It occurs due to mutations in the ATP7B gene, which encodes the liver copper-transporting ATPase protein. A characteristic symptom of Wilson's disease is the accumulation of copper in various organs and tissues, mostly in the liver and basal ganglia.
Radiation hypoparathyroidism
Radiation hypoparathyroidism occurs as a result of radiation damage to the pancreas during external irradiation of the head and neck organs, as well as endogenous irradiation during the treatment of diffuse toxic goiter or thyroid cancer with radioactive iodine.
DiGeorge syndrome
DiGeorge syndrome is a genetic disease caused by impaired embryonic development of the third and fourth pharyngeal pouches.
The incidence of DiGeorge syndrome is 1: 3000–20,000. Such a significant discrepancy in data is due to the fact that a reliable and clear boundary between this disease and velocardiofacial syndrome has not yet been established. The difference between DiGeorge's disease is severe immunological disorders, which are mild in velocardiofacial syndrome.
The genetic nature of DiGeorge syndrome is damage to the central part of the long arm of chromosome 22 (22q22.3), where genes encoding the processes of embryogenesis are presumably located. One of these genes is TBX1, which produces the T-box protein. Evidence of the relationship between DiGeorge syndrome and TBX1 is the fact that a small percentage of patients do not have significant damage to chromosome 22, but only mutations in this gene [15].
Many manifestations of DiGeorge syndrome are detected immediately after the birth of the child. Most often, the first to be detected are anomalies of facial development - cleft of the soft and hard palate, sometimes in combination with a “cleft lip”, prognathism of the lower jaw. Children with DiGeorge syndrome are often short, have a short nose with a wide bridge, deformed or underdeveloped ear cartilages, and hypertelorism. With a relatively mild course of the disease, all of the above symptoms can be expressed rather weakly; even a cleft of the hard palate can occur only in the posterior part and can only be detected during a thorough examination by an otolaryngologist.
The result of PTG hypoplasia is PTH deficiency and persistent hypocalcemia, resulting in the development of a convulsive syndrome, which can manifest itself in the first hours of life (neonatal tetany). This pathology is combined with a wide variety of defects of the cardiovascular system - tetralogy of Fallot, ventricular septal defect, patent ductus arteriosus and others.
The syndrome is characterized by complete or partial underdevelopment of the thymus gland, where cellular immunity is formed. Lymphocytopenia is detected in the blood. T-lymphocyte deficiency is manifested by a tendency to viral, fungal and bacterial infections, which often take a protracted and severe course.
Barakat syndrome
Barakat syndrome, or HDR syndrome (hypoparathyroidism, deafness, renaldisease).
This is a rare genetic disease associated with a defect in the GATA3 (10p15) gene. The gene encodes a protein responsible for the development of the prostate gland, inner ear, and kidneys [16]. Hypocalcemic afebrile seizures and tetany may occur after birth or at any age. PTGs are small endocrine formations, the PTH content is sharply reduced or undetectable. Patients exhibit hypocalcemia, involuntary muscle contractions (tetany), or seizures in infancy. Bilateral sensorineural hearing loss can range from mild to profound. Renal anomalies include dysplasia, hypoplasia, aplasia, and glucocorticoid-resistant nephrotic syndrome (renal failure leading to the loss of large amounts of protein in the urine).
Kenny–Caffee syndrome
Kenny-Caffee syndrome is an orphan disease known as acrocephalopolysyndactyly (ACPS). Inherited in an autosomal dominant manner. The syndrome is based on mutations in the TBCE gene (1q43–44), which cause changes in cell structure. Patients are born with intrauterine growth retardation. Congenital anomalies affect several body systems. Clinically, this is manifested by proportional dwarfism, a small face and convex forehead, acrocephaly (a high conical skull due to premature fusion of cranial sutures), syndactyly or polydactyly.
Other symptoms include visual abnormalities and episodes of tetany associated with hypoPT. The bone cavities and canals of all bones are narrowed (medullary stenosis) with a normal or thickened cortex. Blood tests reveal hypocalcemia and hyperphosphatemia [17].
Kenny-Caffee syndrome type 2
Kenny-Caffee syndrome type 2 (KCS2) is an extremely rare skeletal disorder caused by a mutation in the FAM111A gene, with an autosomal dominant pattern of inheritance.
Children are born short and with low body weight. The head is hydrocephalic, the forehead is convex, microphthalmia. Characteristic is thickening of the tubular bones and osteosclerosis.
Hypocalcemic seizures can be detected already in the neonatal period, which are associated with hypoPT [18].
Sagnada–Sakati syndrome
Sagnada-Sakati syndrome (HRD syndrome, hypoparathyroidism/retardation/dysmorphism) is also known as acrocephalopolysyndactyly type III. This syndrome is caused by a defect in the 1q.42-q.43 gene. Although Sagnada-Sakati syndrome shares the same locus with the autosomal recessive form of Kenny-Caffee syndrome, patients with the latter have normal intelligence and skeletal features.
The syndrome is characterized by intrauterine growth retardation and microcephaly. The face is elongated, narrow, eyes are deep-set, the nose is beak-shaped, the ears are large and drooping, the lips are thin, the bridge of the nose is flat, micrognathia, long filter. Mental retardation is mild or moderate.
In addition, with Sagnada-Sakati syndrome, the development of the skeletal system is disrupted: deformities of the arms and legs, short fingers (brachydactyly), polydactyly, underdevelopment of the lower leg bones and abnormalities of the femurs. Children are born with congenital heart and eye defects. Some were found to have defects in the formation of blood vessels (tortuosity of retinal vessels, superior mesenteric artery), and corneal opacity.
Congenital hypoPT manifests itself early as tetany or increased convulsive readiness. Patients are significantly stunted [19].
Read the end of the article in the next issue
V. V. Smirnov1, Doctor of Medical Sciences, Professor P. N. Vladimirova
Federal State Budgetary Educational Institution of Russian National Research University named after. N. I. Pirogova Ministry of Health of the Russian Federation, Moscow
1 Contact information
Hypoparathyroidism in childhood and adolescence (part 1) V. V. Smirnov, P. N. Vladimirova For citation: Attending physician No. 6/2018; Page numbers in the issue: 49-53 Tags: thyroid gland, endocrine diseases, target organs, calcium
Seizures
The main symptom of hypoparathyroidism is frequent attacks of titanic cramps in the limbs.
Convulsive syndrome is characterized by a sudden development, but gradually patients note a number of signs indicating the onset of muscle spasms in the arms or legs:
- feeling cold;
- tingling feeling in the muscles;
- “goosebumps” in the area between the nose and upper lip;
- muscle numbness and stiffness.
In most cases, cramps affect the limbs symmetrically, but muscle tissue of the entire torso, face and even internal organs can also be affected.
Convulsive syndrome in hypoparathyroidism has some features of manifestation, depending on the area affected by spasms:
- muscles in the arms - the upper limbs are pressed towards the body, involuntary flexion of the elbows and wrists occurs;
- muscle tissue on the face - the eyebrows move, the eyes close, the corners of the lips droop and the jaw clenches;
- coronary vessels - an angina attack occurs;
- core muscles - sharp extension of the body backwards;
- abdominal muscles (intercostal, cervical or diaphragm) - breathing difficulties begin, shortness of breath occurs;
- smooth muscles of the gastrointestinal tract - symptoms of dysphagia and intestinal colic are observed;
- bladder muscles - leads to temporary anuria.
Seizures in other pathologies usually do not cause pain. With hypoparathyroidism, the patient feels unbearable pain, which in severe cases can last for hours. In mild forms of the pathology, convulsions occur no more than twice a day and last no longer than 10 minutes.
How does the disease progress in children?
Endocrine diseases affect not only adults, but also children. Pathologies of the parathyroid gland can occur even in a newborn child. The clinical picture of hypoparathyroidism in childhood is somewhat different. In addition, the disease in children carries a greater health risk.
In children with hypoparathyroidism, an autoimmune form is often detected. There is no systematic pattern of seizures. a harbinger of spasms is severe pain in the abdomen, after which cramps (tonic) occur and the child throws his head back. The patient has difficulty breathing and develops cyanosis of the skin.
If attacks occur frequently, the pressure inside the skull begins to increase, which causes persistent headaches, and as a result, swelling of the optic nerve.
In young children, teething may be delayed, the teeth themselves are deformed, and the enamel is not sufficiently developed.
With hypoparathyroidism in childhood, concomitant diseases can develop:
- autoimmune keratoconjunctivitis;
- cataract;
- disorders of the gonads of the primary type;
- alopecia areata or total;
- lymphocytic chronic thyroiditis;
- chronic adrenal insufficiency;
- Addison–Beermer disease.
Lack of treatment threatens to increase the intensity of symptoms, causing suffering, and also leads to complications. In children with long-term progression of hypoparathyroidism, mental and physical underdevelopment is diagnosed.
Classification
Hypoparathyroidism is classified according to its clinical course. There are several forms of pathology that have characteristic signs of convulsive attacks.
Forms of hypoparathyroidism.
1. Chronic. Brief seizures are uncommon. They are provoked by physical overload of muscles, infectious processes, nervous overstrain, psychological trauma or menstruation. Seasonal exacerbations are noted. It is difficult to cure completely, but long-term remission can be achieved.
2. Acute. The patient experiences frequent and prolonged convulsions, which are very intense and are accompanied by other symptoms of hypoparathyroidism. The patient's condition stabilizes with great difficulties.
3. Latent. It also has a name - hidden, due to the absence of external symptoms of the disease. The cause of muscle spasms can only be determined through diagnosis.
Hypoparathyroidism is also classified according to causative factors.
1. Post-traumatic. It includes idiopathic factors, autoimmune disorders, infections, radiation therapy, internal hemorrhages, etc.
2. Postoperative. The cause of this type of disease is complications after a previous operation on the parathyroid or thyroid gland.
3. Congenital. It develops in the womb or in the first months of a child’s life due to underdevelopment or complete absence of the parathyroid glands.
The patient's seizures may have a certain character. For example, in some patients spasm occurs only in the lower or upper extremities, while in others only in the internal organs.
Treatment of hypoparathyroidism
Treatment of the disease is primarily aimed at stopping the attack - tetany requires prompt assistance. Long-term therapy involves restoring calcium and magnesium metabolism and, in some cases, taking hormonal medications. Treatment is combined with diet therapy and vitamin therapy. If CNS dysfunction is observed, then therapy is aimed at supporting the nervous system. It is recommended to combine taking medications with physiotherapeutic procedures.
The attack is stopped as quickly as possible. This is necessary due to the high risk of complications caused by circulatory disorders in the coronary vessels. The patient may suffer from asphyxia during an attack. Timely therapy involves the administration of calcium supplements. Most often, a 10% calcium salt solution is used. Depending on the volume of the injected solution, the effect lasts from 2 to 8 hours. Emergency assistance can only be provided by medical professionals or persons who have undergone special training. The introduction of calcium chloride and calcium gluconate has its own characteristics. The volume is determined according to the patient's condition. Constant monitoring is necessary to avoid overdose.
In addition to intravenous administration of solutions, oral administration of medications is possible. In addition to calcium, drugs containing parathyroid gland extract are often prescribed. They allow you to maintain natural control of calcium levels in the blood - that is, they perform the function of endogenous parathyroid hormone. Such medications are always prescribed during therapy with calcium salts.
The basis of long-term therapy is taking calcium and magnesium supplements to restore normal metabolism. As a rule, they start with large dosages, gradually reducing them in accordance with the improvement of the condition. This dosage regimen allows you to quickly relieve the symptoms of the disease and ensure normal functioning. Subsequently, an individual maintenance dosage is selected for each patient. It is important to take calcium supplements together with vitamin D - it improves the absorption of calcium in the intestines.
It is recommended to take the medicine after meals - the active substance has an irritating effect on the gastric mucosa. At the same time, drugs that bind phosphorus are used - these are almagel or aluminum hydroxide. In addition to calcium, magnesium sulfate is taken. During therapy, it is necessary to control blood pressure.
More about the consequences
Despite the fact that hypoparathyroidism leads to disruption of metabolic processes and functions of internal organs, the prognosis for recovery is favorable if therapy is started in a timely manner. However, hypoparathyroidism requires not a single, but a course of treatment, including systematic examinations, medication and clinical observation.
The patient should visit the endocrinologist 1 - 3 times within 3 months. If the disease is completely eliminated, then a visit to the doctor can be made once every six months. In addition to the endocrinologist, you should see an ophthalmologist, since hypoparathyroidism causes disturbances in the condition of the visual organs. Hypoparathyroidism leads to cataracts in 60% of cases.
If the pathology has not been diagnosed for a long time and no action has been taken to eliminate it, the following consequences may be observed:
- emotional lability;
- serious sleep disturbances;
- decreased intellectual capabilities;
- excessive dryness of the skin followed by peeling scales;
- hyperpigmentation of a large area of the skin;
- different types of eczema;
- development of fungal dermatitis;
- constant fragility of the nail plates;
- partial/complete baldness.
Hypoparathyroidism is dangerous not only because of the consequences that occur after any period of time, but also because of the complications that can arise during an attack. Muscle spasms can lead to tissue atrophy, and chest cramps can lead to suffocation.
Our doctors
Mikhailova Elena Vladimirovna
Endocrinologist, diabetologist, candidate of medical sciences
Experience 46 years
Make an appointment
Slovesnova Tatyana Alekseevna
Endocrinologist, Candidate of Medical Sciences, doctor of the highest category
Experience 48 years
Make an appointment
Kolodko Inna Mikhailovna
Doctor - endocrinologist, doctor of the highest category
Experience 26 years
Make an appointment
Hypocalcemic crisis
A sudden and sharp decrease in calcium levels during hypoparathyroidism leads to a hypocalcemic crisis. The patient's condition is deteriorating greatly and requires immediate medical attention.
An external irritant can provoke a crisis, and the condition also worsens spontaneously. Severe convulsive attacks can affect the smooth muscles of the bronchi and vocal cords, resulting in broncho-or laryngospasms, which provokes severe respiratory failure. The crisis can last from 10 to 15 minutes, up to 3 or more hours, if no therapy is carried out.
Hypocalcemic crisis is preceded by:
- cold hands or feet;
- severe weakness;
- impaired sensitivity of facial tissues and fingers at the tips;
- panic;
- fibrillary twitching, turning into convulsions.
It is difficult to cope with a hypocalcemic crisis on your own. Without medical help, an attack can have serious consequences. In addition, the longer the muscles are affected by spasms, the greater the likelihood of complications.
Diagnostics
As a rule, patients with complaints of seizures initially turn to a therapist. The doctor interviews the patient about symptoms and also studies the patient’s medical history. The physician may order general laboratory tests to determine preliminary results. After receiving basic information about the clinical picture, the therapist sends the patient to an endocrinologist.
The specialist conducts a more detailed survey about the nature and all the features of seizures, possible causes and duration of the disease. If the patient has problems with the endocrine system and has been treated, you should definitely inform the doctor about these facts.
Initially, the patient is given general recommendations for stopping attacks and diagnostic methods are prescribed. Until the results are received, the specialist cannot establish a diagnosis and prescribe full treatment.
Diagnosis of hypoparathyroidism includes:
- analysis of hormones in the blood;
- analysis of the concentration of minerals (calcium and phosphorus) in urine;
- consultation and examination by an ophthalmologist;
- X-ray to identify possible lime deposits;
- MRI - helps to determine calcium deposits in the brain, on the external tissues of internal organs and subcutaneous tissue;
- densitometry - identifying a possible increase in indicators of increased bone tissue density.
The latent form of the disease is more difficult to establish. The absence of symptoms in lateral hypoparathyroidism suggests that the diagnosis is complicated, so the doctor may prescribe, in addition to the listed examination methods, differential diagnosis and hyperventilation tests.
Modern diagnostic methods make it possible to identify hypoparathyroidism at the initial stage of its development, which facilitates and shortens the treatment process, and also reduces the risks of complications. Particular attention should be paid to children for whom hypoparathyroidism, even with moderate severity, is dangerous with serious consequences.
Recommendations
Preventing hypoparathyroidism is not easy. The main method of prevention is timely treatment of any diseases of the endocrine system and the elimination of factors that negatively affect it.
Regarding hypoparathyroidism, experts recommend that if it is necessary to perform surgery on the thyroid gland, not to refuse surgical intervention, and, if possible, to carry out treatment using minimally invasive methods.
Endocrinologists distinguish a disease such as toxic recurrent goiter. When repeating treatment, it is better to carry out radioactive iodine therapy instead of another surgical intervention.
In the postoperative period, you must strictly adhere to all prescriptions and actively engage in the prevention of endocrine pathologies.
The disease develops in the following cases:
- if surgery was performed on the organs of the neck, as a result of a disorder of the parathyroid glands and thyroid gland. Hypoparathyroidism can occur if the entire thyroid gland has been removed (thyroidectomy) or if cancer is detected;
- if hemorrhages occur in the parathyroid gland due to neck injuries;
- formation of inflammation in the parathyroid gland;
- when tumors appear in the neck and parathyroid glands;
- in the presence of a congenital pathology (the parathyroid glands are underdeveloped in utero) - manifests itself in DiGeorge syndrome, its characteristic features are a violation of the parathyroid glands, congenital heart defects, thymic aplasia;
- the effect of radiation (during treatment with radioactive iodine, toxic goiter);
- for endocrine disorders (chronic adrenal insufficiency, primary hypothyroidism);
- for diseases amyloidosis and hemochromatosis, for autoimmune syndrome.