The prevalence of neurofibromatosis type 1 (NF1) is approximately 1 case in 3,000 people, but this figure varies markedly depending on the country and in Russia it is 1 case in 7,812 people. The life expectancy of people with neurofibromatosis type 1 is reduced by ~8–21 years, and their cause of death is most often related to malignancy.
NF1 is a genetically determined disease with an autosomal dominant pattern of inheritance, which means that all carriers of germline mutations in the 17q11.2 gene will develop NF1. However, the manifestations of this disease are extremely variable and can differ even among members of the same family who have an identical mutation. By the way, there are quite a lot of different mutations that cause the disease (>3000), which is associated with the large size of the gene (more than 60 exons) and the variety of types of mutations (translocations, deletions, inversions and point mutations) that can occur in this area - this complicates genetic diagnosis. The latter is carried out on DNA material from peripheral blood lymphocytes and/or tumor material, with the exception of segmental neurofibromatosis (sometimes called neurofibromatosis type V), when the lesion affects only one segment of the patient’s body. In the latter case, only tumor material can serve as diagnostic material. .
Figure 1 | Segmental neurofibromatosis. Multiple neurofibromas located within one or more dermatomes.
The 17q11 gene encodes the protein neurofibromin, which is a tumor suppressor. Neurofibromin is produced in nerve cells and specialized neuroglial cells (oligodendrocytes and Schwann cells) and normally suppresses the RAS proto-oncogene, accelerating its transition to an inactive form. The inability to produce functional neurofibromin results in increased cell growth and survival.
General information
Neurofibromatosis is a group of inherited, relatively rare diseases that present with a variety of symptoms and phenotypic variability.
Most often, neurofibromatosis (NF) is manifested by characteristic pathological changes - the development of tumors predominantly of ectodermal origin with damage to the skin, the central nervous system, the presence of specific pigment spots on the skin with a “coffee with milk” color, abnormalities in the development of the bone skeleton and a number of other manifestations. ICD-10 code for neurofibromatosis: Q85.0 (non-malignant neurofibromatosis). Neurofibromatosis is the most common form of hereditary monogenic pathology, the incidence of which varies from 1:2000 to 1:40000. The disease is characterized by an autosomal dominant type of inheritance with a frequency of manifestation (penetrance) approaching 100%. There were no differences in the incidence of lesions by gender.
In general, based on clinical and morphological manifestations, eight types of neurofibromatosis have been identified and described, the most significant of which are:
- Neurofibromatosis type 1 (NF 1). About 90% of all neurofibromatosis are of type NF1, which is the most common among diseases of this group.
- Neurofibromatosis type 2 (NF 2). It occurs much less frequently (1:40,000), but has a more aggressive course than NF1.
Some authors believe that of the eight forms of neurofibromatosis, all except NF2 are considered abortive and should not be distinguished as independent nosological forms.
Neurofibromatosis type 1 (synonymous with Recklinghausen's disease)
The frequency of occurrence in newborns is 1:4000, in the Russian population - 1.28:10,000. Transmission along the paternal side is more typical than through the maternal side. In more than half of the cases, the manifestations of the disease are minimal; incomplete and monosymptomatic forms are often found. About 80% of neurofibromatosis cases are sporadic and result from new mutations. Due to the high frequency of mutations inherent in the NF1 gene, which determine the clinical development of neurofibromatosis, it is assumed that either its extremely high mutability or the presence of mutations in several loci of the gene.
Neurofibromatosis type I is characterized by pronounced polymorphism of clinical manifestations, a progressive course and frequent involvement of various organs and systems in the process with the development of severe complications, often leading to death. Recklinghausen's disease (neurofibromatosis) has a clear tendency to progress slowly. There are two periods of sharp increase in the activity of the pathological process: the first from 5 to 10 years and the second from 35 to 50 years. At the same time, the second period of activity in the overwhelming majority of cases (about 75%) is due to malignancy of tumor neoplasms. Provoking factors that contribute to the growth of neurofibromas include conditions and age-related changes accompanied by hormonal changes in the body: pregnancy , puberty.
A feature of the disease is the specific sequence of manifestation of the symptoms of neurofibromatosis type 1, depending on the patient’s age, which causes difficulties in its diagnosis in young children. Thus, neurofibromatosis in children in the first years of life/from birth can be present only in the form of some signs of the disease, more often these are large pigment spots or skeletal dysplasia and plexiform neurofibromas , and other characteristic symptoms can manifest much later (by 5-15 years). And if we take into account that the degree of their severity, the course and rate of progression of the pathological process vary widely, the difficulties in making a diagnosis become understandable, which contributes to its late diagnosis.
Neurofibromatosis type 2
Neurofibromatosis type II occurs independently of NF-I; its manifestations are determined by the localization of the main pathological process and corresponds to the symptoms of damage to the brain/spinal cord. In the spinal cord, the process often involves the membranes of the spinal roots of the cervical and lumbar regions in the form of Schwannoma (a benign tumor of Schwann cells of the nerve sheaths), cauda equina (dorsal roots); when localized in the brain, the process may involve the cranial nerves, medulla oblongata, and pons. Less commonly, peripheral nerves are involved in the process with the development of ependymomas and meningiomas .
The average age of manifestation of signs of the disease in NF 2 is 20 years, and at the time of diagnosis - about 28 years. Typically, NF 1 begins to manifest in early childhood, primarily with skin symptoms, while NF 2 begins at a young age, primarily with the development of deafness due to the development of vestibular schwannomas and other symptoms secondary to spinal schwannomas and meningiomas.
Brain stem gliomas
They make up approximately 8–10% of the total number of CNS neoplasms in NF1. The average age of patients is 8 years. The cerebellum is most often affected, followed by the pons and diencephalon. Concomitant hydrocephalus and brain herniation should be noted. Tumors appear hyperintense on T2WI and usually exhibit contrast enhancement heterogeneously. To avoid missing low-intensity contrast enhancement, subtraction may be used. .
Figure 7 | Brain stem glioma. In the area of the cerebellum, a hyperintense formation is detected on T2WI (a), which on post-contrast T1WI (b) demonstrates weak inhomogeneous accumulation of contrast.
Pathogenesis
The pathogenetic changes in neurofibromatosis type 1 are based on the high frequency of spontaneous mutations of the NF1 gene (17q11.2), which encodes the cytoplasmic tumor suppressor protein neurofibromin, which is produced in nerve and specialized neuroglial cells. Neurofibromin contains a special domain, which exerts a suppressive effect on the proliferation of cells predominantly of neuroectodermal origin due to the inactivation of the adapter protein Ras, which is an inhibitor of the structure of mitogenic signaling pathways. With this kind of genetic defect in chromosomes 17q, the dynamic balance of growth regulation is disrupted and it is shifted towards proliferation and the formation of benign tumor growth.
More than 500 types of mutations in the gene on chromosome 17q have been described, which interfere with the regulatory function of the NF1 gene in oncogenesis. The mechanism of development of clinical manifestations is still unknown. It is generally accepted that the pathology is based on the development of various tumors ( neurinoma , neurofibroma ) from the nerve sheaths. About half of the cases are the result of new mutations.
The genetic defect in neurofibromatosis type 2 is located on chromosome 22 (22q12) and directly encodes the process of synthesis of the Merlin protein, which is also a tumor suppressor. It is of greatest importance in regulating the proliferation (reproduction) of cells of neuroectodermal origin. With a mutation, the synthesis of Merlin in one chromosome does not manifest itself at the cellular level, and with a symmetric mutation (damage) of the allelic gene in the cell, the synthesis of Merlin stops and, accordingly, the balance inherent in a healthy body in the regulation of growth shifts (drifts) towards proliferation with the development of the process of benign tumor growth .
Medicines
Photo: prostatit.guru
Medicines are used in the treatment of neurofibromatosis, but their purpose is only for symptomatic treatment; they do not eliminate the problem itself. The following medications are used:
- ketotifen;
- fenkarol;
- tigazol;
- lidase.
These drugs are prescribed by a doctor strictly individually, since the use of these drugs is not rational in every case.
Tigazol contains vitamin A. There are practically no contraindications to the use of the drug; people with liver or kidney failure, as well as in cases of individual intolerance to the component, should be careful.
Ketotifen is an antiallergic drug and has a membrane-stabilizing effect. Fenkarol also belongs to antiallergic drugs. The drug has a good antipruritic effect, therefore it is prescribed to people who have itching in the clinical picture of the disease. Fenkarol is well tolerated and causes virtually no side effects. Contraindicated for people with gastrointestinal and kidney diseases. It should be remembered that the drug does not have a depressant effect on the central nervous system, but in rare cases a mild sedative effect is observed.
Lidase contains hyaluronidase as an active substance. This is an enzyme whose action is aimed at hyaluronic acid. The drug increases the permeability of tissues, improves their trophism, increases the elasticity of scar areas, promotes the resorption of hematomas and eliminates contractures.
Symptoms
Recklinghausen's neurofibromatosis begins to manifest itself in most cases as multiple neurofibromas , localized along the peripheral nerves, in the form of round painful nodules located in the thickness of the skin, varying in size. Neurofibromas are presented in the form of nodules on/in the thickness of the skin of normal or bluish-red color and soft elastic consistency.
A characteristic feature, starting from the neonatal period/first years of life of children, is the appearance on the skin of small “coffee spots”, reminiscent of freckles with clear boundaries and smooth edges, with their predominant localization in areas of the body with folds (in the groin area, armpits, on the abdomen) . As the child grows older, there is a tendency for their number (from single spots to several hundred) and sizes (from pinpoint to 2-5 centimeters in diameter) to increase; they can also acquire a dark color. It should be taken into account that diagnostically significant spots include in children before puberty - with a diameter of more than 5 mm and after puberty - 15 mm or more. Neurofibromatosis (photos of the initial stage) are given below.
Soft skin tumors, as a rule, are absent in children, especially in the first years of life and appear by 10-15 years. At the same time, their number especially increases during puberty. hypoesthesia may occur . Other changes may be present on the skin: areas of depigmentation, vascular spots, focal graying of hair, hypertrichosis . Development of neurofibromas . Plexiform neurofibroma can form giant tumors consisting of cutaneous and subcutaneous elements.
Some authors (V.V. Mordovtseva) identify four variants of the clinical course of neurofibromatosis type I:
- with the presence of minor pigment spots such as freckles in combination with large “coffee” spots in the absence of neurofibromas or with the presence of single tumors;
- with the presence of predominantly large pigmented coffee spots on the skin and a small number of neurofibromas;
- with a predominance of neurofibromas and a small number of pigment spots (like freckles/large ones);
- mixed type.
Optional symptoms of neurofibromatosis are manifested by changes in the skeletal system in the form of various deformities of the spinal column - scoliosis , lordoscoliosis , kyphoscoliosis , pseudarthrosis , thinning/curvature of long tubular bones, pathological fractures. Facial dysmorphia often develops: anomalies of the palpebral fissures, ocular hypertelorism , deformation of the auricles, irregular shape of the skull, etc. Patients with NF-I are characterized by macrocephaly , short stature, dysplasia of the sphenoid bone and occipital part of the skull.
Characteristic lesions of the nervous system are convulsive and hypertensive-hydrocephalic syndromes, tumors of the central nervous system ( ependymoma , astrocytoma , glioma of the optic nerve/brain stem), epilepsy , which appears with corresponding symptoms. Thus, optic nerve glioma manifests itself as visual impairment.
NF1 is characterized by additional manifestations in the form of mild to severe cognitive impairment, often in combination with a moderate decrease in the child’s IQ, difficulty in learning to read, write, and mathematics, and neuropsychic imbalance; the presence of endocrine disorders (puberty, pheochromocytoma , growth disorders, etc.). On the vascular side, patients often experience renal artery stenosis with increased blood pressure, arterial occlusion , and Moya-Moya disease .
Clinical manifestation of NF2 disease includes the development of bilateral vestibular schwannomas or schwannomas of other cranial peripheral and spinal nerves with minimal extraneural and cutaneous symptoms. Bilateral schwannoma (neurinoma) of the auditory nerve is one of the main symptoms, which occurs in almost 90% of cases, is rarely found in children, and develops mainly in late adolescence/early adulthood. NF-II.
meningiomas (spinal, intracranial, optic nerve meningiomas), gliomas and epindymomas occur . The first symptom is often decreased/loss of hearing, the appearance of noise in the ears, which may be accompanied by ataxia and dizziness. Cafe au lait spots on the skin are in most cases absent or present in small quantities. In 50-70% of patients with neurofibromatosis type 2, visual disturbances ( hemarthromas , cataracts, retinoblastomas , optic nerve meningiomas ) are diagnosed.
The table below shows the characteristic manifestations of neurofibromatosis types 1 and 2.
Folk remedies
Photo: ambafra-pk.org
Traditional medicine will never get rid of neurofibromatosis, but can help with complex treatment prescribed by a doctor. Despite the fact that folk remedies are considered harmless, before using any recipes, it is recommended to consult a doctor for advice, who will tell you all the benefits and possible harms of a particular remedy.
To maintain immunity, do not forget about nutrition. You should increase the content of fermented milk products (milk, kefir, fermented baked milk), seafood (sea fish, seaweed) in your diet; among meat products, preference is given to chicken and beef; green tea is also useful, as it has an antioxidant effect. It is also recommended to get rid of bad habits. This will help strengthen the general condition of the body.
There is a recipe for an infusion of celandine. To prepare it you need to take 1 tbsp. l. herbs and pour three glasses of boiling water. After infusion, you should drink one glass 15 minutes before meals. This plant is considered poisonous in high concentrations, so it is important to maintain proportions when preparing the infusion. It should be noted that the prepared infusion is stored for no more than 48 hours; after this time, consuming the infusion is strictly prohibited.
Burdock infusion is also used. Both stems and roots of the plant are suitable for its preparation. These ingredients are brewed in water at the rate of 30 g per 1 liter of water. After preparation, the infusion is used 3 times a day.
The information is for reference only and is not a guide to action. Do not self-medicate. At the first symptoms of the disease, consult a doctor.
SEARCH FOR TREATMENT AROUND THE WORLD WITH YELLMED
Symptoms of neurofibromatosis in children
The symptoms of neurofibromatosis type 1 in children are similar, but the manifestation of characteristic signs is closely tied to the age of the child. Below is a table of characteristic signs of neurofibromatosis type I and complications in children at different age periods (Wikipedia).
The most common manifestation is “coffee spots” on the child’s skin. Below is a photo of neurofibromatosis in children.
In NF1 children, coordination of movements may be slightly impaired and muscle tone may be reduced. Also, many children have a larger skull size than their peers (exceeding more than 4 standard deviations). They may also be stunted. Such children often have little initiative and are emotionally limited compared to healthy peers. Almost 12% of children with NF1 have a characteristic phenotype: antimongoloid eye size, hypertelorism (increased distance between paired organs, such as the eyes), low-set ears, pulmonary stenosis, cervical pterygum. NF-II is found extremely rarely in children.
Treatment
Often, the management of patients with neurofibromatosis is limited to symptomatic treatment. If neurofibromas are located in areas of increased trauma and cause pain, they are removed surgically. In case of multiple formations, chemotherapy is prescribed and radiation is performed. If the musculoskeletal system is damaged, rehabilitation measures are carried out.
The disease has a favorable prognosis. Tumor formations become malignant extremely rarely. When performing special measures, patients maintain a satisfactory quality of life and performance.
Consequences and complications
Complications are extremely varied in nature and include:
- Malignization (malignant transformation) of tumors.
- Decreased vision/blindness.
- Development of pheochromocytoma .
- Essential arterial hypertension .
- Renal artery stenosis.
- Vasculitis/vasculopathy with damage to the cerebral/coronary and arteries. Compressive neuropathy .
- Deformities of the spinal column/chest - scoliosis , lordoscoliosis , kyphoscoliosis , pseudarthrosis .
- Thinning/curvature of long tubular bones, pathological fractures.
- Cosmetic defects.
- Cognitive impairment (attention deficit disorder).
Skeletal abnormalities
Normal functioning of bone tissue requires a coordinated interaction between resorbing (osteoclasts) and bone-forming (osteoblasts) cells. The use of a mouse model showed impaired osteoblast function (increased pyrophosphate production, decreased bone morphogenetic protein 2, which promotes osteoblast differentiation) in mice mutant for the NF1 gene. As a result, patients experience impaired bone mineralization, arched deformities of the tibia, kyphotic and scoliotic deformities, chest deformities, pseudarthrosis of large joints, and dysplasia of the sphenoid bones.
Forecast
The prognosis in each specific case is individual and is largely determined by the severity of the disease and the rate of its progression. Life expectancy in patients with neurofibromatosis is similar to the average in the population or is reduced by an average of 15% with the development of severe complications or tumor malignancy. By obtaining the appropriate level of education, patients with rational employment can lead a normal life. depressive syndrome due to a gross cosmetic defect .
Focal areas of hyperintensity (FASI)
Found in approximately 80% of patients with NF1, they are areas of hyperintense MR signal on T2WI and FLAIR located in the basal ganglia, thalamus, brainstem, cerebellum and subcortical white matter. Morphologically they represent areas of myelinopathy with enlarged vacuoles. The correlation of these changes with the clinic is not fully understood. .
Figure 8 | Asymmetric areas of hyperintense MR signal in T2 are detected in the basal ganglia on both sides. Also noteworthy is the hyperintense formation on the left surface of the neck and head - plexiform neurofibroma.
List of sources
- Sergeev A. S. Mutation frequency of neurofibromatosis // Abstract of thesis. Ph.D. biol. Sci. M., 1973 (USSR Academy of Medical Sciences, Institute of Medical Genetics).
- Schneider N. A. Neurofibromatosis of the first type: Recklinhausen disease / N. A. Schneider, A. I. Gorelov //Siberian Medical Review. - 2007. - No. 3 (44). — P. 91-95.
- Kozlov A. V. Neurofibromatosis 2 (NF2) // Surgery of skull base tumors / Edited by A. N. Konovalov. - M.: OJSC "Mozhaisk Printing Plant", 2004. - P. 169-170. — 372 s
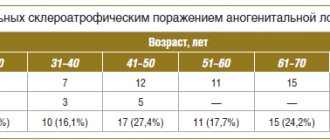
- Ufimtseva M.A., Bochkarev Yu.M., Galperin A.M., Golovyrina I.L., Gurkovskaya E.P. SKINAL MANIFESTATIONS OF RECKLINGHAUSEN'S DISEASE // Modern problems of science and education. – 2021. – No. 6.
- Popova A. A Clinical and diagnostic aspects of neurofibromatosis // University Medicine of the Urals - 2021 - No. 2.