Diagnostics
If the patient does not have the main symptoms of purpura (bleeding and hemorrhage), doctors have difficulty establishing the correct diagnosis.
You need to be especially careful not to confuse purpura with vascular skin abnormalities. Purpura is diagnosed, as a rule, according to the clinical and hematological picture. First, a hematologist examines the patient. To confirm the diagnosis, the doctor prescribes the patient to undergo the following hardware examinations and tests:
- general clinical and biochemical blood tests;
- urea test;
- general clinical urine test;
- myelogram.
Also, to establish purpura, differential diagnosis is carried out by identifying symptoms of diseases such as hemolytic-uremic syndrome, hemolytic microangiopathic purpura and hepatorenal syndrome.
As a rule, purpura is treatable, but there are cases of death if the bleeding and hemorrhage are very heavy.
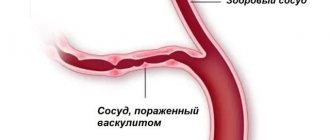
Hemorrhagic vasculitis (Henoch-Schönlein disease)
The disease is usually manifested by a triad of symptoms: hemorrhagic skin rashes (purpura), arthralgia and (or) arthritis (mainly of large joints) and abdominal syndrome, which is observed in almost 2/3 of patients. If the last two signs are present, an increase in body temperature is usually observed.
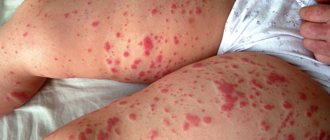
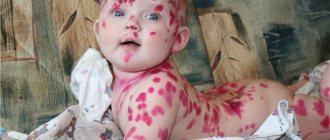
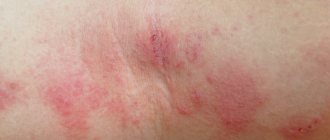
Initial skin rashes are erythematous, sometimes itchy papules located on the extensor surfaces of the extremities, most often on the legs, buttocks, and rarely the torso. Subsequently, the papules turn into typical purpura, going through all stages of development up to hyperpigmentation, which persists for a long time. In severe cases, areas of necrosis develop that become crusted.
Joint damage is observed in more than 2/3 patients. Large joints are usually involved in the process. Patients can only be bothered by arthralgia of varying intensity - from aches to acute pain leading to immobility, or polyarthritis caused by periarthritis and synovitis. Characterized by volatility and symmetry of the lesion.
Abdominal syndrome (abdominal purpura) usually develops in children; it is characterized by pain similar to intestinal colic, localized around the navel, but often in other parts of the abdomen - in the right iliac region, right hypochondrium, epigastrium, simulating appendicitis, cholecystitis, pancreatitis. At the same time, patients experience a typical picture of abdominal syndrome - pale skin, sunken eyes, pointed facial features, dry tongue, symptoms of peritoneal irritation. Patients usually lie on their side, huddled, with their legs pressed to their stomach, or toss and turn in bed. Abdominal pain may be mild, without a specific localization, and patients remember them only when questioned. Simultaneously with colic, bloody vomiting and loose stools appear, often streaked with blood. Palpation of the abdomen always increases pain, revealing signs of damage to the peritoneum. All the variety of abdominal purpura can be summarized in the following options:
- typical intestinal colic;
- abdominal syndrome simulating appendicitis or intestinal perforation;
- abdominal syndrome with the development of intussusception.
Patients with abdominal syndrome should be observed by a physician and a surgeon so that, if necessary, the issue of surgery can be resolved in a timely manner.
Not so rarely (usually in patients with abdominal syndrome) the kidneys are involved in the pathological process, and hematuric glomerulonephritis develops due to damage to the glomerular capillaries. According to the severity of the pathological process, glomerulonephritis can occur in a variety of ways - from isolated urinary syndrome to diffuse glomerulonephritis of hypertensive or mixed type, rarely with nephrotic syndrome. Typically, a favorable course of glomerulonephritis is observed; chronic progressive nephritis with renal failure is possible.
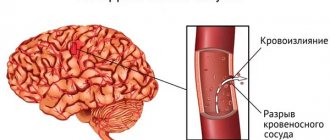
Clinical signs of central nervous system damage , hemorrhagic pneumonia, myocarditis and serositis are rarely observed and are recognized using special research methods.
Treatment
The essence of the treatment of purpura is to reduce the amount of production of antiplatelet antibodies and make it impossible for them to bind to platelets.
If the patient experiences bleeding, the doctor prescribes a course of hemostatic drugs, as well as aminocaproic acid. If the bleeding is also uterine, then the patients are prescribed oxytocin. In the event that there is a risk of hemorrhage in the brain, surgeons prescribe platelet infusion (infusion) to the patient.
There is also a surgical method for treating purpura. In this case, the patient undergoes surgery to remove the spleen (splenectomy). But it is very important to remember that surgical interventions are an extreme case when conservative therapy is ineffective.
Another method of treating purpura is plasmapheresis, or a procedure for purifying blood plasma from antibodies and toxins by filtering the blood plasma using special devices.
Doctors also advise patients with vascular purpura to follow a hypoallergenic diet.
Henoch-Schönlein purpura
Henoch-Schönlein purpura (hemorrhagic vasculitis) is a systemic vasculitis affecting the microvasculature (arterioles, capillaries and post-capillary venules), with typical deposition in the vascular wall of immune complexes consisting of immunoglobulin A (IgA). Clinically, the disease manifests itself as a hemorrhagic skin rash, joint syndrome, damage to the gastrointestinal tract (GIT) and kidneys.
Henoch-Schönlein purpura develops at any age, but the maximum incidence is observed in children aged 4-6 years, amounting to approximately 13-18 cases per 100 thousand. With age, the incidence decreases and the development of the disease after 60 years is considered rare.
The etiology of the disease has not been established, however, an increase in the incidence of Henoch-Schönlein purpura in the cold season, as well as the frequent association of the onset of the disease with episodes of acute respiratory or intestinal infection may indirectly indicate the infectious nature of the disease. The list of etiological agents associated with the development of Henoch-Schönlein purpura includes β-hemolytic streptococcus group A, Haemophilus influenzae, chlamydia, mycoplasma, Legionella, Yersinia, Epstein-Barr, Coxsackie, hepatitis B and C viruses, adenovirus, cytomegalovirus, parvovirus B19 , salmonella, Helicobacter pylori, Clostridium difficile. There are isolated observations of Henoch-Schönlein purpura that developed after vaccination against typhoid fever, measles, and influenza. Alcohol, medications, food products, hypothermia, and insect bites can be triggers for the disease.
Pathogenesis
Currently, Henoch-Schönlein purpura is considered an immune complex disease associated with the deposition of granular IgA deposits in the vascular wall and activation of complement. This concept is based on the results of numerous studies showing impaired synthesis and/or metabolism of IgA in the majority of patients with Henoch-Schönlein purpura: increased levels of serum IgA, IgA-containing immune complexes, and IgA-fibronectin complexes. However, the pathogenetic significance of these disorders requires further evaluation. In recent years, evidence has accumulated indicating the anti-inflammatory properties of IgA, which gives grounds to regard the increase in its synthesis as a compensatory process that occurs secondarily in response to an inflammatory reaction in the mucous membranes. Thus, it has been shown that IgA has the ability to reduce the production of proinflammatory cytokines and is not able to activate complement; IgA is found in the endothelium of unaffected vessels and in the mesangium of unchanged renal glomeruli; The observation of Henoch-Schönlein purpura with complete selective IgA deficiency is described. Considering the frequent association of the development of Henoch-Schönlein purpura with episodes of infections of the respiratory tract and gastrointestinal tract, this assumption seems quite probable.
Another reason for changes in IgA metabolism in Henoch-Schönlein purpura may be a violation of O-glycosylation of the hinge region of the heavy chains of the IgA1 molecule, which, as has been shown, can lead to impaired clearance of IgA1 by liver receptors and prolongation of the circulation period of IgA polymers and IgA-containing immune cells. complexes in the systemic circulation. It has been shown that IgA1 molecules with aberrant glycosylation acquire the ability to activate complement via an alternative pathway and have an increased tropism for the mesangial matrix of the renal glomeruli.
In recent years, additional data have been obtained that indirectly confirm the assumption about the infection-dependent nature of Henoch-Schönlein purpura. Thus, it was shown that in the majority of patients during an exacerbation of cutaneous vasculitis, transient endotoxemia is observed - circulation of lipopolysaccharide of gram-negative bacteria in the systemic bloodstream. The pathogenetic significance of this phenomenon in Henoch-Schönlein purpura requires further study, but the possibility of endotoxin involvement in the development of vascular inflammation mediated by the Schwartzman reaction is assumed. Chronic inflammation of the intestinal wall, possibly caused by dysfunction of its local immune system or an infectious process, can play an important role in the pathogenesis of endotoxemia. This assumption is supported by the discovery of increased intestinal permeability for macromolecules (ovalbumin) in the majority of patients with Henoch-Schönlein purpura during exacerbations of cutaneous vasculitis. In addition, the presence of a chronic inflammatory process in the mucous membrane of the small intestine in patients with Henoch-Schönlein purpura has been demonstrated, which, apparently, is the morphological basis for dysfunction of the intestinal barrier and the development of transient endotoxemia.
Clinical picture
The clinical picture of Henoch-Schönlein purpura consists of four typical manifestations: hemorrhagic skin rash, damage to the joints, gastrointestinal tract and kidneys. In most cases, the disease develops gradually, gradually and does not significantly affect the general condition of the patients. As a rule, this variant of the onset of the disease is observed with isolated skin lesions. The number of organ manifestations of Henoch-Schönlein purpura varies from 1-2 to a combination of all 4 classical signs, which can develop in any order over several days or weeks of illness. In some cases, in addition to the mentioned manifestations, damage to other organs, in particular the lungs, heart, and central nervous system, may develop.
Skin lesions are observed in all patients with Henoch-Schönlein purpura and are a mandatory diagnostic criterion. In most cases, a hemorrhagic rash is the first clinical manifestation of the disease, which is subsequently accompanied by damage to other organs and systems. The most typical localization of skin rashes: lower extremities - legs and feet. Often the skin rash spreads to the thighs, buttocks, torso, upper limbs and extremely rarely to the face. In the process of evolution, hemorrhages gradually become pale, transform into brown pigment spots and then disappear. With a long-term recurrent course, the skin in the affected area may become pigmented due to the development of hemosiderosis. In most cases, the hemorrhagic rash is represented by petechiae and purpura, but in some cases erythematomacular and urticarial elements may also be observed.
Joint damage, as a rule, develops in parallel with skin damage and occurs as migratory polyarthralgia, less often - arthritis. The favorite localization is the knee and ankle joints; the elbow, wrist and other joints are less commonly affected. These manifestations of the disease are always transient and benign, never leading to the development of permanent changes in the joints.
Damage to the gastrointestinal tract is observed in 60-80% of childhood patients and 40-65% of adult patients. The most constant symptom: abdominal pain, worsening after eating, which often creates a typical picture of “abdominal toad.” A common complication of abdominal lesions in Henoch-Schönlein purpura is intestinal bleeding.
Kidney damage in Henoch-Schönlein purpura can become chronic and is the main factor determining the prognosis of the disease as a whole. The incidence of renal involvement varies from 30 to 70% depending on the age of the patients. In adults, kidney damage is detected almost 2 times more often than in children. As a rule, clinical signs of kidney damage are detected in the first 3 months of the disease, however, with a chronic recurrent course of cutaneous vasculitis, delayed appearance of signs of glomerulonephritis is possible - several months or even years after the onset of the disease. Possible predictors of renal involvement in children include male gender, age over 5 years, abdominal syndrome, persistent cutaneous purpura, and decreased plasma levels of factor XIII. In adult patients, risk factors for kidney damage include the presence of fever and episodes of infections at the onset of the disease, the spread of skin rash on the torso, severe abdominal syndrome and the presence of laboratory signs of inflammatory activity of the disease. The severity of renal pathology, as a rule, does not correlate with the severity of skin manifestations of the disease, however, in both children and adults, a significant positive correlation was noted between the frequency of kidney damage and the development of abdominal syndrome, which requires more careful dynamic monitoring of the corresponding cohort of patients. In children, in half of the cases, kidney damage has a favorable course with complete clinical and laboratory recovery, while in most adult patients there is a tendency towards a chronic persistent course of nephritis.
In half of patients with Henoch-Schönlein purpura, glomerulonephritis is manifested by microhematuria, which, as a rule, is combined with minimal or moderate proteinuria. A third of patients experience gross hematuria, which most often develops at the onset of nephritis, but can also occur at later stages of renal damage during exacerbations of cutaneous vasculitis or respiratory infections. More severe types of renal damage are also possible, including nephrotic syndrome, rapidly progressive nephritis and acute renal failure. Arterial hypertension syndrome is detected in 14-20% of patients. The development of chronic renal failure (CRF) as a result of glomerulonephritis is observed in 12-30% of patients.
Diagnostics
Diagnosis of Henoch-Schönlein purpura is based on identifying typical clinical signs of the disease, primarily bilateral hemorrhagic skin rashes at the time of examination or in the medical history. There are no specific laboratory tests for Henoch-Schönlein purpura. Changes in the clinical blood test - an increase in the erythrocyte sedimentation rate (ESR) - may reflect the inflammatory activity of the disease, as well as the severity of complications (anemia due to intestinal bleeding). The presence of thrombocytopenia is a criterion for excluding Henoch-Schönlein purpura. A marked increase in ESR and significant dysproteinemia are not typical for Henoch-Schönlein purpura. Disease activity is reflected by the level of von Willebrand factor and thrombomodulin in the blood plasma. The detection of high levels of fibrin/fibrinogen degradation products in plasma in active forms of the disease is not a sign of the development of DIC syndrome, but only reflects the high inflammatory activity of the disease. The examination plan for all patients must include a virological and immunological blood test to exclude other diseases that occur with skin purpura.
A key role in confirming the clinical diagnosis is played by a biopsy of the skin and/or kidneys, less often of other organs, with the obligatory immunohistochemical study revealing the fixation of IgA-containing immune complexes in the vascular wall. It should be taken into account that in addition to Henoch-Schönlein purpura, IgA deposits are found in skin lesions as part of chronic inflammatory bowel diseases (Crohn's disease, ulcerative colitis), chronic diffuse liver diseases of alcoholic etiology, celiac disease, and Dühring's dermatitis herpetiformis.
The morphological picture of kidney damage in Henoch-Schönlein purpura is identical to that in Berger's disease (primary IgA nephropathy). The most common morphological variant of kidney damage is mesangioproliferative glomerulonephritis, characterized by focal or diffuse proliferation of mesangiocytes. Immunohistochemical examination reveals granular deposits of IgA, less often IgG, as well as the C3 component of complement, fibrin. In more severe cases, the formation of epithelial “crescents” is noted.
Widely used classification diagnostic criteria for Henoch-Schönlein purpura, proposed in 1990? American College of Rheumatology and including the patient's age less than 20 years, palpable purpura, abdominal syndrome and the morphological picture of cutaneous leukocytoclastic vasculitis (2 or more of 4 criteria are required), have little practical significance due to their low sensitivity and specificity (87.1 and 87. 7%, respectively).
Differential diagnosis
Differential diagnosis is carried out with a wide range of diseases that occur with damage to small vessels:
- primary vasculitis of small vessels (Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, cryoglobulinemic vasculitis). The results of blood tests for antineutrophil cytoplasmic antibodies (Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome) and cryoglobulins (cryoglobulinemic vasculitis) have differential diagnostic significance; morphological examination data (granulomatous inflammation in Wegener's granulomatosis, eosinophilic vasculitis in Churg-Strauss syndrome). Of particular importance is the immunohistochemical examination of biopsy specimens of affected tissues. Detection of IgA deposits is a characteristic sign of Henoch-Schönlein purpura.
- Vasculitis in autoimmune diseases (systemic lupus erythematosus, rheumatoid arthritis, Sjogren's disease, Crohn's disease, ulcerative colitis). Differentiation is based on the clinical features inherent in each nosology, the results of laboratory and instrumental research methods.
- Vasculitis in infections (subacute infective endocarditis, tuberculosis, infection with hepatitis B and C viruses), malignant neoplasms, drug allergies.
Treatment
For skin lesions, the following drugs from the sulfonamide group can be effective: sulfasalazine (orally, 500-1000 mg 2 times a day), colchicine (orally, 1-2 mg once a day). Glucocorticoids are effective in the vast majority of patients, especially in high doses, but their long-term use in patients with Henoch-Schönlein purpura without involvement of internal organs is undesirable, since the severity of side effects in such a situation may exceed the severity of the disease itself. The prescription of non-steroidal anti-inflammatory drugs (NSAIDs) is justified only in cases of severe articular syndrome and the ineffectiveness of other drugs. In other cases, prescribing NSAIDs should be avoided due to the adverse effect on the intestinal mucosa and increased intestinal permeability.
Damage to the gastrointestinal tract with intense abdominalgia is an absolute indication for the use of glucocorticoids:
prednisolone intravenous drip 300-500 mg/day for 3 days in a row, followed by switching to oral administration 0.5 mg/kg 1 time per day for 2-3 weeks, then quickly reducing the dose by 5? mg every 3 days to full cancellations.
Gastrointestinal bleeding (if it is caused by vasculitis and not other reasons) is not a contraindication to the administration of oral glucocorticoids, but, on the contrary, serves as one of the main indications for such treatment. A contraindication to the administration of glucocorticoids orally for abdominal syndrome can only be perforation of the intestinal wall, which currently extremely rarely complicates the course of Henoch-Schönlein purpura.
The greatest problems in the drug treatment of Henoch-Schönlein purpura are associated with the choice of treatment for chronic glomerulonephritis. Most authors consider the use of ultra-high doses of glucocorticoids, cytostatics and/or plasmapheresis sessions to be justified in cases of severe glomerulonephritis (nephrotic syndrome with normal or impaired renal function; more than 50% of glomeruli with epithelial “crescents”).
In this case, the following scheme is used:
- prednisolone orally 1 mg/kg 1 time per day for 4-6 weeks, then reduce the dose by 2.5 mg/week until complete withdrawal or prednisolone intravenous drip 15 mg/kg 1 time per day for 3 consecutive days (total 6-20 three-day “pulses” with an interval of 3-4 weeks); +
- cyclophosphamide intravenous drip 15 mg/kg once every 3-4 weeks, under the control of the level of peripheral blood leukocytes and transaminases (6-20 “pulses” in total); +
- plasmapheresis with an exfusion volume of 30-60? ml/kg, 10-14 sessions.
Single uncontrolled studies have shown the effectiveness of a combination of glucocorticoids and azathioprine in severe cases of kidney damage, as well as a combination of glucocorticoids and cyclophosphamide with antiplatelet agents or anticoagulants.
In addition, for the treatment of patients with nephrotic and rapidly progressive glomerulonephritis, it is proposed to use intravenous immunoglobulins:
normal human immunoglobulin intravenously at 400-1000 mg/kg for 1-5 days, repeated courses once a month for 6 months.
There is no consensus regarding less severe forms of glomerulonephritis. With isolated microhematuria, minimal proteinuria (up to 0.5 g/day) and preserved renal function, as a rule, active immunosuppressive treatment is not required. For moderate proteinuria (0.5-1 g/day), the prescription of drugs that affect the non-immune mechanisms of progression of kidney damage is indicated: angiotensin-converting enzyme inhibitors and/or angiotensin II receptor antagonists (due to their ability to reduce intraglomerular hypertension and the severity of proteinuria), statins (for lipid metabolism disorders). Some retrospective studies have shown a beneficial effect of tonsillectomy on the course of mild forms of glomerulonephritis in Henoch-Schönlein purpura.
Correction of hemostasis disorders, previously considered a priority in the treatment of Henoch-Schönlein purpura, is currently considered only as an auxiliary method of therapy, the prospects of which are assessed with skepticism. In practical terms, the reports of Japanese researchers on the favorable long-term clinical and pathomorphological effect of fibrinolytic therapy with urokinase on the course of glomerulonephritis in Henoch-Schönlein purpura are of interest:
urokinase intravenously slowly 5000 IU/kg 3 times a week for 3-12 weeks.
According to the authors, the action of urokinase may be based on reducing the severity of intraglomerular hypercoagulation and dissolving fibrinogen/fibrin deposits.
Forecast
An important clinical prognostic factor that determines the incidence of chronic renal failure is the severity of proteinuria. So, if with minimal proteinuria chronic renal failure develops in 5% of patients, then with nephrotic syndrome this figure increases to 40-50%. The most unfavorable with regard to the development of chronic renal failure is the combination of nephrotic syndrome with arterial hypertension and impaired renal function at the onset of glomerulonephritis.
The most important morphological criterion for determining the prognosis of kidney damage is the proportion of renal glomeruli with “crescents” from the total number of glomeruli. Thus, according to French authors who observed 151 patients from 1 year to 18 years, in the presence of “crescents” in more than 50% of the glomeruli, end-stage renal failure developed in 37% of patients, and in another 18% glomerulonephritis had a chronic progressive course. On the other hand, 85% of patients who achieved end-stage renal disease had crescents in more than half of the glomeruli. In 70% of patients with complete recovery or minimal changes in urine tests, no “crescents” were found in the glomeruli.
It is important that the majority of patients with late progression of glomerulonephritis do not have clinical signs of activity of renal and extrarenal lesions, which is explained by the predominant influence of non-immune mechanisms of progression on the course of renal damage. In this regard, in all patients with Henoch-Schönlein purpura with kidney damage, careful monitoring of blood pressure and correction of metabolic disorders, in particular hyperuricemia and dyslipidemia, are extremely important.
Why does rheumatic purpura appear?
Hemorrhagic vasculitis, also called Henoch-Schönlein disease, is a type of inflammation of small areas of the vascular system.
A similar process can occur in the human body due to failures in the stable functioning of the immune system. The main sources of disease development include:
- Acute and chronic diseases caused by bacterial or viral infection (for example: acute respiratory viral infections, staphylococci, herpes rashes, etc.)$
- Hereditary predisposition;
- Poisoning of the body with biological poisons and chemicals;
- Prolonged exposure to frost or heat;
- Insect bites;
- Personal reaction to the introduction of special types of vaccine preparations;
- Sunburn of the skin.
When should you see a doctor?
If you begin to worry about the problems presented below, then urgently make an appointment with a specialist:
- A sudden rash in the form of crumbs in the areas of the arms, legs, hip area and face;
- Regular pain and discomfort in the joints, in particular this concerns the lower extremities of the body;
- Edema and swelling;
- The appearance of bruises;
- The presence of blood in the urine;
- A sharp rise in pressure.