General information
Hemophilia - what is this disease?
Hemophilia (Latin - haemophilia) is a genetically inherited disease based on a hereditary deficiency (decreased activity) of plasma coagulation factors (VIII - hemophilia A or IX - hemophilia B), manifested by hemorrhagic syndrome (decreased blood clotting, a pronounced tendency to hemorrhage). bleeding). Blood clotting factor refers to a protein found in platelets/blood plasma that allows blood to clot. Normally, the level of blood clotting factor activity varies between 50-150%.
Type of inheritance: X-linked recessive and in the vast majority of cases (about 70%) there is a positive family history. Hemophilia is caused by mutations in the FIX (Xq27) or FVIII (Xq28) gene. Less common are cases without a family history due to sporadic mutations (Wikipedia). A sporadic gene mutation can occur during ovulation/spermatogenesis in any generation and remain hidden for several generations of female carriers until the X chromosome carrying the hemophilia gene is accidentally passed on to a boy. Moreover, in spermatogenesis , mutations in the gene occur 4-5 times more often than in oogenesis . That is, in 80-85% of sporadic cases of the disease, mothers are carriers of a gene mutation that arose in the germ cells of the father. There is a strong correlation between the age of the father and the risk of his daughter receiving a mutation from him: the average age of the father is 40 years.
Hemophilia (incoagulability of blood) is a fairly rare disease (annual incidence rates in the human population vary between 7-15 cases/100,000 population). At the same time, hemophilia A (HA) accounts for 78-88% of the total number of cases of the disease. In the vast majority of cases, hemophilia affects males. Women are conductors (carriers/conductors) of the hemophilia gene and pass on parts of this disease to their sons.
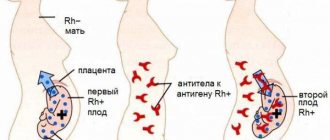
Isolated cases of hemophilia also occur in females when the gene is inherited from a father with hemophilia and from a carrier of the gene - the mother, or in women with a gene mutation on one chromosome and an inactive gene on the other chromosome ( Shereshevsky-Turner disease ). It is extremely rare that some women who carry clotting factor (FIX)/coagulation factor (FVIII) gene mutations may have clinical manifestations.
Below is a diagram of the inheritance of hemophilia, according to which the probability of having sons without signs or with signs of hemophilia in women who are carriers of the pathological gene and in a healthy man is the same (50:50). At the same time, some daughters may have symptoms of the disease, and the probability of having a daughter who carries the gene is 50%. A man with hemophilia will be able to conceive healthy children with a woman who is not a carrier of the pathologically altered gene. Moreover, all born daughters will be carriers of the disease.
In addition to genetically determined (hereditary) forms, acquired forms of hemophilia (Rosenthal disease) are distinguished, which are based on factor XI deficiency and which belongs to the group of rare coagulopathies . Acquired forms of hemophilia are extremely rare and their development is based on the appearance of antibodies to coagulation factors, which is typical for autoimmune/myeloproliferative diseases, hypersensitivity to drugs, skin diseases and pregnancy . Acquired hemophilia is most common in the age period of 69-80 years, but among women the pathology often occurs during reproductive age. Overall, the prevalence of acquired hemophilia is 1.5 cases/1 million population/year (Figure below).
Often in the literature,
hemophilia A is called the “Victorian” or “royal disease” disease, which appeared in Russia thanks to the descendants of Queen Victoria and from which the son of Nicholas 2, Alexey, suffered. The ill-fated gene was inherited by the child’s mother (Alexandra Fedorovna, who was Victoria’s granddaughter), which she passed on to her son. The spread of hemophilia among the reigning persons of Europe was also facilitated by marriages between close relatives, practiced with the aim of strengthening dynasties and political expediency, examples of which are well known in the history of certain monarchies.
Pathogenesis
The pathogenesis of hemophilia is based on a deficiency of plasma factors (VIII, IX, XI) of blood coagulation, which causes a disruption of the process of blood coagulation in the internal coagulation unit of hemostasis (formation of thromboplastin) and causes hematoma delayed type of bleeding. The first stage of blood coagulation is the process of thromboplastin formation, which occurs normally only if there is a sufficient concentration of factors VIII and IX. Its duration is 12-15 minutes, and the subsequent process of blood clotting after the appearance of active thromboplastin in the blood occurs almost instantly.
It is the disruption of this plasma phase of hemostasis that causes the type of bleeding characteristic of hemophilia. Bleeding immediately after injury may be absent, since primary (vascular-platelet) hemostasis functions normally, which ensures the formation of a primary thrombus.
Since the primary thrombus is not able to provide a final stop of bleeding, secondary hemostasis suffers - the formation of a fibrin (final) thrombus, the bleeding resumes and occurs suddenly a few hours (the next day) after surgery/trauma. At the same time, despite its duration, there is no increase in the volume of blood loss, which is its feature. The figure below schematically shows the pathogenesis of hemophilia.
Gastrointestinal bleeding
In the case of severe hemoglobin and severe deficiency of coagulation factors (more often with a combined deficiency of factors VIII and IX), traces of blood can be noticed already during the first regurgitation after feeding the baby. Excessively hard food and children swallowing small objects (especially those with sharp edges or protrusions) can injure the mucous membrane of the gastrointestinal tract.
As a rule, if fresh blood is detected in the vomit or during regurgitation, then damage should be looked for on the mucous membrane of the esophagus. If the vomit looks like “coffee grounds,” then the source of the bleeding is in the stomach; the blood has had time to react with hydrochloric acid, and hydrochloric acid hematin has formed, which has a characteristic appearance.
If the source of the bleeding is in the stomach, there may also be the presence of black, often liquid, tarry stools called melena. If there is fresh blood in the child's stool, bleeding from the lowest parts of the intestine - the rectum and sigmoid colon - can be suspected.
Classification
There are several types of disease:
- hemophilia A - caused by hereditary deficiency of antihemophilic globulin (factor VIII);
- hemophilia B (syn. Christmas disease) - based on deficiency of Christmas factor (blood clotting factor IX);
- hemophilia C is a rare variant of hemophilia caused by factor XI deficiency;
- hemophilia A + B is an extremely rare variant (occurrence 1-1.5%), in which there is a combined deficiency of factors VIII and IX.
Classification of hemophilia A and B - according to the severity of the disease, determined by the activity of FVIII and FIX, according to which 3 forms of severity are distinguished:
- Mild (blood clotting factor activity varies between 5-50%). In general, severe bleeding is rare, mainly in cases of major trauma/surgery.
- Moderate severity (blood clotting factor activity within 1-5%). Spontaneous bleeding and severe bleeding during surgery/trauma are possible. Hemorrhagic syndrome is moderate, develops mainly in preschool age, exacerbations 2-3 times a year.
- Severe form (clotting factor activity less than 1%). It is characterized by the development of severe hemorrhagic syndrome, spontaneous post-traumatic bleeding mainly into muscle tissue/joints, less often into internal organs, diagnosed in 60-70% of cases. In children it is accompanied by the development of severe hemorrhagic syndrome at an early age.
What it is?
Hemophilia is a hereditary blood disorder that is caused by a congenital absence or reduction of blood clotting factors. The disease is characterized by a blood clotting disorder and manifests itself in frequent hemorrhages in the joints, muscles and internal organs.
This disease occurs with a frequency of 1 case per 50,000 newborns, with hemophilia A diagnosed more often: 1 case of the disease per 10,000 people, and hemophilia B less often: 1: 30,000-50,000 male residents. Hemophilia is inherited through a recessive trait associated with the X chromosome.
In 70% of cases, hemophilia is characterized by a severe course, steadily progresses and leads to early disability of the patient. The most famous hemophiliac in Russia is Tsarevich Alexei, the son of Alexandra Feodorovna and Tsar Nicholas II. As you know, the disease was inherited by the family of the Russian emperor from his wife’s grandmother, Queen Victoria. Using the example of this family, the transmission of the disease along the genealogical line is often studied.
Causes
The direct cause of the development of hemophilia A/B is gene mutations of the X chromosome in the area of the long (q27-q28) arm. About 70-75% of patients have a family history of hemorrhagic syndrome (in relatives), and in 25-30% the inheritance of the disease in the family history is not traced, that is, spontaneous gene mutation occurs on the X chromosome.
It was revealed that the gene that encodes the process of factor VIII synthesis is located in the Xq 28 locus on the long arm of the X chromosome and consists of 25 introns/26 exons. The size of the factor VIII gene is 186 thousand paired DNA bases. Moreover, mutations in this gene can occur in different types: deletions, duplications, inclusions of new bases, reading frame shifts. In most cases, an inversion of the nucleotide sequence is observed, in particular, an inversion in the factor VIII gene of intron 22, which causes a low level of this factor and also causes severe phenotypic manifestations.
The factor IX gene is located on the long arm of the X chromosome at locus Xq 27 and includes 7 introns/8 exons. At the same time, mutations in the factor IX gene occur 7-10 times less frequently than in the factor VIII gene. In severe hemophilia, two types of gene mutations often occur.
Combined clinical syndromes
The genes encoding the development of color blindness and hemophilia are located at a very close distance on the X chromosome, so cases of their joint inheritance are not uncommon. At the birth of a daughter, the disease does not develop into a clinical form, but in 50% of cases the daughter becomes a carrier of the pathological gene.
A boy born in the marriage of a colorblind man and a healthy woman has a 50% chance of being colorblind, but if the mother is a carrier of a defective gene and the father is sick (colorblindness or colorblindness along with hemophilia), then the birth of a sick boy is 75%.
Symptoms
The degree of clinical manifestations and blood coagulation disorders depends on the level of factor activity in the blood, but in clinical practice, a direct correlation between the clinical phenotype of the disease and laboratory data does not always exist. Clinical symptoms of hemophilia are manifested by hemorrhagic syndrome, manifested in the form of hemarthrosis , which accounts for 70-80% of the manifestations of hemophilia ; hematomas and bleeding . Frequent recurrence of hemorrhagic syndrome is typical, and its distinctive feature is the inadequacy of the severity of the injury.
Hemarthrosis
Hemarthrosis (bleeding in the joints) is a typical manifestation of the disease, and the source of bleeding in the joint is predominantly the joint capsule. In the absence of timely and adequate treatment, bleeding may continue until the joint is completely filled with blood. In young children, joint hemorrhages are rare. However, as the child grows up and motor activity increases, the load on the joints increases, their frequency increases sharply. The most commonly observed hemarthrosis is the knee (48%), elbow (24%) and ankle (16%) joints; the shoulder/hip (5-8%) and small joints of the feet/hands are much less commonly affected. Cases of damage to the joints of the spine are extremely rare. As a rule, depending on the severity of the disease/patient’s age, several joints are affected (from 1 to 8-10).
Hemorrhage in the joints develops mainly for no apparent reason. Characteristic is a rapid increase in joint volume, local hyperemia / hyperthermia , severe pain and tension of the skin over the joint. The figure below shows the clinical symptoms of hemarthrosis, depending on the volume of hemorrhage into the joint cavity.
There is a dysfunction of the joint, a contracture is formed: movements in the affected joint are limited, the joint is deformed and, when straightening the limb, takes on irregular shapes.
After the first hemorrhages, as a rule, the blood in the synovial cavity slowly and gradually resolves, and the function of the joint is also gradually restored. However, if hemorrhage in a certain joint occurs frequently, then the reaction of gradual development of arthritis , and such a joint is usually called the “target joint”.
With frequent hemorrhages in the joint and the absence of prompt/adequate replacement therapy, hemophilic arthropathy , which is a persistent deforming change in the joints. Gradually, arthrosis affects the cartilage and joint capsule, and later the pathological process spreads to the soft tissues/adjacent bones. The inflamed synovial membrane gradually thickens, and local vessels are easily damaged, which is the source of repeated hemorrhages. Developing fibrosis of the capsule/surrounding tissue deforms the joint and severely limits its mobility. Under the influence of proteolytic enzymes, the cartilage of the joint is destroyed (cartilage degeneration), and osteoporosis with the formation of cysts filled with gelatinous fluid.
The musculature surrounding the joint gradually atrophies, which increases the load on the joint. In hemophilic arthropathy, the following forms of hemarthrosis are distinguished: acute hemarthrosis (primary, recurrent); posthemorrhagic synovitis (acute, subacute, chronic); deforming osteoarthritis ; ankylosis (fibrous/osseous). As a rule, the early occurrence of hemarthrosis contributes to persistent damage to the surfaces of the joints and its components, and the development of multiple arthropathy leads to profound disability of the patient (Fig. below).
Hematomas
Another characteristic manifestation of hemophilia includes hematomas of various locations (intermuscular, subcutaneous, subperiosteal, retroperitoneal, subserous, etc.), which are characterized by a pronounced tendency to spread. The spilled blood remains liquid for a long period, which facilitates its easy penetration into adjacent tissues and rapid spread along the fascia. The process of resorption of hematomas is slow.
The greatest threat to the patient's life is represented by retroperitoneal hematomas , the symptoms of which resemble the clinical picture of acute appendicitis . The cause of partial intestinal obstruction can be subserous hematomas, which often inhibit the wall and contents from breaking into the intestinal lumen. Also, extensive hematomas in some cases can compress large arteries/nerve trunks, causing circulatory disorders and impaired sensitivity. Moreover, compression of blood vessels by the hematoma can cause tissue necrosis and the formation of “hemophilic pseudotumors.”
Clinically, the process of hematoma formation can be accompanied by both local (pain, sensation of heat, tingling/numbness, limitation of movement due to muscle spasm) and general symptoms (weakness/malaise, loss of appetite/sleep, fever during resorption of the hematoma). Also, when blood is shed in large quantities, anemia can develop.
Bleeding
In patients with hemophilia, bleeding is easy and frequent, it is profuse and prolonged, and can occur at any time of the day. The frequency of bleeding is largely determined by the severity of hemophilia. Thus, the frequency of bleeding episodes in a patient with severe hemophilia A can vary within the range (15-35 times/year or more). In patients with moderate/mild forms of hemophilia A and B, bleeding develops much less frequently. Traditional measures to stop bleeding are ineffective.
There are external bleedings (from the mucous membranes of the mouth, gums, nose), which occur in 75% of cases, and internal bleeding (renal, gastrointestinal, hemorrhages in the brain/meninges), which are relatively rare and occur mainly after various instrumental manipulations/ as a result of injuries. Prolonged renal bleeding occurs in 14-30% of patients with severe hemophilia, while hematuria often occurs spontaneously and can lead to the development of anemia . As a rule, the development of internal bleeding in most patients is accompanied by acute pain. There are two categories of bleeding, the criterion of which is the degree of danger to life: life-threatening and ordinary.
Life-threatening bleeding includes bleeding into vital organs. Their frequency varies between 5-10%, but they often cause paralysis/death (hemorrhages in the spinal canal, meninges, brain, gastrointestinal tract). Retropharyngeal bleeding is especially dangerous , since due to the accumulation of blood clots in the upper respiratory tract it can cause suffocation.
Typically, the severity of hemorrhage does not correspond to the intensity/extensiveness of the injury, and hemorrhages may occur delayed (6-12 hours to 24 hours) after injury depending on the degree of factor deficiency. This is due to the fact that the basis for the primary control of bleeding is the vascular-platelet component of hemostasis, the functions of which are not impaired in hemophilia.
Tests and diagnostics
The diagnosis of hemophilia is made on the basis of a study of family history, physical examination (the patient has manifestations of cutaneous hemorrhagic syndrome in the form of multiple hematomas/ecchymoses of varying severity), clinical manifestations (joint damage in the form of swelling/deformation, local increase in skin temperature, disorders range of motion/joint mobility, limb muscle wasting, etc.), as well as laboratory coagulological examination data (activated partial thromboplastin time, thrombin time, prothrombin time, blood clotting time, blood fibrinogen concentration according to Clauss, decreased level of factor VIII/IX, etc. ). Molecular genetic research may also be performed.
In order to assess the severity of joint damage and the condition of organs in which profuse bleeding is noted (kidneys, stomach, intestines), an X-ray examination, ultrasound/CT/MRI of joints/soft tissues, chest and abdominal organs, urinary tract, brain, esophagus are performed -gastroduodenoscopy/colonoscopy.
Most often, the diagnosis of hemophilia is made in early childhood, when the child begins to walk and parents note that his blood does not clot.
The average age of diagnosis of hemophilia, the severe form, is 9 months; for moderate forms - 22 months, and mild forms of the disease are diagnosed in children at a later age, mainly after invasive interventions/tooth extraction. A screening test for disorders of the coagulation system is the clotting time, and when this indicator is prolonged, studies of the level of factors in the blood are carried out, on the basis of which the diagnosis of “hemophilia” is established and the severity of the disease is determined.
Questions and answers
Is it possible to be completely cured of hemophilia?
Drug therapy and blood or plasma transfusion procedures relieve acute pathology syndromes. Sometimes patients are prescribed long-term use of medications selected based on medical history. But completely eliminating the symptoms of hemophilia remains impossible.
Does hemophilia pose a threat to a child's life?
If parents seek medical advice in a timely manner, the baby will be out of danger. A quick correct diagnosis and initiation of treatment will allow the child not to limit himself in physical activity and games.
Can a boy with hemophilia pass it on to his sons?
The risk of having children with hemophilia from a father with bleeding problems is minimal. The disease will be inherited by sons only if their mother is one of the carriers of the altered gene.
Hemophilia in children
The acquired form of hemophilia in children is very rare. Congenital forms in children are much more common. In newborns, severe hemophilia is manifested by hemorrhages in the brain and bleeding from the remnant of the umbilicus. Immediately after birth, only a severe form appears - extensive cephalohematoma and extensive hemorrhages in the tissue after injections. According to the degree of danger in newborns, bleeding is arranged in descending order: cerebral, intramuscular, into the mucous membranes and skin. Cerebral hemorrhages occur in 10% of newborns, and they can be confirmed by ultrasonography.
Cerebral bleeding becomes dangerous if a hemophilic history is not established before birth. Mild and moderate forms are not recognized in infants, but often manifest as prolonged hidden bleeding leading to severe anemia. The average age of diagnosis for severe hemophilia is 8-9 months (extensive hematomas and prolonged bleeding appear), and for an average degree it is 1.5-2 years. The time of appearance of the first symptoms correlates with the severity of the disease: the earlier it appears, the more pronounced the deficiency of a particular factor. In the first year, bleeding during teething may be the first manifestation of the disease. Also at this age, biting the tongue and cheek mucosa is also accompanied by prolonged bleeding.
In children from one year to 1.5 years, the manifestations of the disease are bleeding and hematomas. Children at this age begin to move actively and the risk of injury and bruising on the forehead, arms, legs and buttocks increases. However, bleeding into the joint is rare before the age of three. After 3 years, due to a more active way, the most typical symptom may appear - hemorrhage in the joint and muscle atrophy .
In younger children, bleeding from the urinary tract rarely occurs, and over the years this symptom occurs more and more often. Gastrointestinal bleeding is also rare. Mild and moderate forms appear only during operations or tooth extraction. In older children, hemarthrosis and intramuscular hematomas after injury and spontaneously. Depending on the severity, the symptoms of the disease may vary. In severe cases, there is spontaneous bleeding that is not related to injury. In mild cases, bleeding occurs after surgery. In moderate hemophilia - with minor trauma and are of a “delayed” nature (time after the injury).
Prevention
Primary prevention of hemophilia involves genetic testing of a married couple whose relatives have cases of hemophilia. Genetic counseling determines the risks of inheriting a gene. If this moment is missed, then a prenatal molecular genetic study is possible, which is carried out at 10-12 weeks (chorionic villus puncture is performed for the study) or from the 15th week (amniotic fluid is taken). If it was not possible to obtain DNA markers, then at week 20 the fetus is taken to determine the level of factor VIII. For early diagnosis, blood is taken from the umbilical cord after the baby is born.
Dispensary observation and treatment of patients
- Carrying out preventive treatment with factor concentrates and “bypass” drugs.
- An examination in specialized hemophilia centers, which evaluates the effectiveness of treatment, drug intolerance, and the presence of an inhibitor to blood clotting factors.
- Treatment of concomitant diseases - teeth, gastrointestinal tract, ENT.
- Treatment of complications of hemophilia ( anemia , factor inhibitors).
- Dentist examinations 2 times a year. During manipulations, the patient must have factor concentrates, Desmopressin or Tranexam .
- Maintaining oral hygiene. Patients should use a soft toothbrush to brush their teeth.
- Patients should avoid intramuscular injections and not use acetylsalicylic acid and other drugs that affect platelet coagulation and function.
- Daily non-strength physical exercises that develop and strengthen muscles are important; swimming and therapeutic walking are useful. At the same time, it is necessary to avoid exercise on weight machines and sports associated with the risk of injury.
Is there a way to cure hemophilia?
Unfortunately, a complete cure for this disease is not yet possible. However, with effective supportive treatments, there is a good chance of improving children's quality of life. Treatment with medications, first of all, includes constant intravenous infusions into the child of the blood clotting component that is missing in his body. Not so long ago, doctors practiced daily administration of such a drug, but thanks to the latest developments in the field of modern hematology, it is now possible to administer the drug much less frequently: once every three days. This makes life much easier for patients.
Now at the laboratory level, specialists are testing other methods of treating hemophilia. The main goal is to replace intravenous injections with oral medications in tablet form. There is also a hypothesis about the possibility of using gene therapy. If it can be made a reality, patients will have the opportunity not to take injections throughout the year.
Consequences and complications
Complications of hemophilia are:
- Hemorrhages in important organs.
- Posthemorrhagic anemia.
- Hemophilic arthropathy ( synovitis , joint deformity). Chronic synovitis appears after several hemorrhages in the joints and is most often associated with the lack of replacement treatment or its insufficient effectiveness. Synovitis is manifested by a local increase in temperature, an increase in joint volume, pain and limited mobility.
- Contractures. osteoarthritis develops with deformation and inflammatory changes in the joints, which ultimately leads to ankylosis and contractures . Deformation and contractures cause disability.
- Fractures. They occur against the background of osteoporosis and arthropathy .
- The emergence of inhibitory antibodies to factors.
- Possibility of transmission of infections (viral, including HIV, prion, parvovirus). When using recombinant factors, this risk is absent.
- Development of pseudotumors. They are formed by hemorrhages in the muscles located with the bone, usually in the muscles if treated incorrectly. They manifest themselves as a tumor-like formation that exists for many months and years and which has no tendency to reverse development, despite the ongoing replacement therapy. The pseudotumor enlarges and compresses surrounding tissues. Without treatment, it reaches very large sizes, creating pressure on nerves and blood vessels, and also causing fractures.
- Complete removal of the tumor and capsule is recommended.
- Nephritis and kidney failure .
- Rheumatoid syndrome as an immune complication.
Forecast
Today, hemophilia throughout the world is not a death sentence, since with early diagnosis and correct replacement treatment, patients live full lives. They can get an education and work, play certain sports, start a family and travel.
Prevention with replacement therapy is an effective way to prevent complications of hemophilia that lead to disability. The main task is to unite doctors and the state in the production of coagulation factors in sufficient quantities to meet the needs of all patients. If at the beginning of the twentieth century. The life expectancy for hemophilia was no more than 13 years, but today, subject to preventive treatment, the life expectancy of patients does not differ from healthy people. At the same time, patients with the inhibitory form are at risk of disability and even death. The greatest risk of developing the inhibitory form in children is observed up to 10 years of age and subsequently decreases.
List of sources
- Protocol for the management of patients with Hemophilia. Problems of standardization in healthcare 2006, p. 18-74.
- Guide to hematology: in 3 volumes. T.3 / Edited by A.I. Vorobyov. 3rd ed. M., 2005.
- Rumyantsev A.G., Rumyantsev S.A., Chernov V.M. Hemophilia in the practice of doctors of various specialties. 2013. 136 p.
- Tretyakova O.S. Hemophilia in children: etiopathogenesis, clinical manifestations, diagnostic approaches //Children's medicine. 2012, 3-4 (16-17) p. 26-35.
- Zozulya N.I., Kumskova M.A., Polyanskaya T.Yu., Svirin P.V. // Clinical guidelines for the diagnosis and treatment of hemophilia. 2021. 34 p.
Hemophilia Society
In many countries of the world, in particular in Russia, special societies have been created for patients with hemophilia. These organizations unite patients with hemophilia, members of their families, medical specialists, scientists studying this pathology and simply individuals who want to provide any assistance to patients with hemophilia. There are websites of these communities on the Internet, where anyone can find detailed information about what hemophilia is, read legal materials on this issue, communicate on a forum with people suffering from this pathology, exchange experiences, ask for advice, and, if necessary, get moral support.
In addition, the sites usually contain a list of links to domestic and foreign resources - foundations, organizations, information sites - with similar topics, giving the visitor the opportunity to comprehensively familiarize himself with the problem of hemophilia or get to know people suffering from this pathology living in his region. The creators of such communities organize special conferences, “schools” for patients, and all kinds of social events in which anyone with hemophilia can take part.
Therefore, if you, your relative or friend suffers from hemophilia, we recommend that you become a member of such a society: there you will certainly find support and help in the fight against such a serious disease.