THALASSEMIA
(Greek thalassa sea + haima blood; synonym:
erythroblastic anemia, target cell anemia, familial microcythemic anemia, hemolytic Mediterranean anemia, hereditary leptocytosis
) - a group of hereditary hemolytic anemias characterized by impaired synthesis of hemoglobin globin chains to varying degrees, as well as hemolysis , hypochromia and ineffective erythrocytopoiesis.
Thalassemias are classified as “quantitative” hemoglobinopathies (see), in which the synthesis of globin chains that are part of the molecules of normal human hemoglobin is reduced, sometimes completely absent (see Hemoglobin); however, the structure of the circuits is not changed.
Due to the great genetic heterogeneity of Thalassemia (the main thalassemic genes (β°, β+, αβ, δβ, etc.), which determines its wide clinical polymorphism, the term “thalassemic syndromes" is sometimes used to designate various forms of Thalassemia. In the classification of Thalassemia based on the results biochemical and genetic studies, the following forms are distinguished: I. Alpha thalassemia (α-thalassemia): 1) heterozygous carriage of the “silent” gene (α-th2), 2) heterozygous carriage of the manifest gene (α-th1), 3) hemoglobinopathy H , 4) homozygous alpha thalassemia (hydrops fetalis with Hb Bart's). II. Beta-thalassemia (β-thalassemia): 1) heterozygous beta-thalassemia, including carriage of the “silent” β-th gene, 2) homozygous beta-thalassemia (Cooley's anemia) - β0- II β+-thalassemia, 3 ) heterozygous δβ-thalassemia (F-thalassemia), 4) homozygous δβ-thalassemia (F-thalassemia) - δβ°- and δβ+-thalassemia. III. Thalassemia associated with structural disorders of hemoglobin: 1) Lepore hemoglobinopathy, 2) Constant Spring hemoglobinopathy (CS).
T. (obviously homozygous beta thalassemia) was first described by Cooley and Lee (Th. B. Cooley, R. Lee) in 1925. Ital. researchers Rietti (F. Rietti, 1925), Greppi (E. Greppi, 1928), Micheli (F. Micheli, 1935) and others reported mild forms of this disease, which later turned out to be heterozygous. Caminopetros (J. Caminopetros, 1938) and Wintrobe (M. M. Wintrobe, 1940) established the hereditary nature of thalassemia. In 1965, JD Heywood et al. Whittroll and Clegg (DJ Weatherall, J.B. Clegg) demonstrated the presence in reticulocytes of patients with beta thalassemia of a partial or complete block in the synthesis of beta polypeptide globin chains in vitro. Alpha thalassemia was described in 1955 by Rigas and Gutta (DA Rigas, A. Gouttas) independently.
T. is distributed mainly in countries located on the coasts of the Mediterranean and Black Seas. Due to population migration, T. genes were brought to America and other parts of the world. Environmental and ethnic factors, consanguineous marriages, and the incidence of malaria in a given area (currently and in the recent past) play a significant role in the spread of T. The highest frequency (up to 30%) of heterozygous carriage of beta thalassemia was detected in Italy. populations (Southern Sardinia). In certain regions of Greece (the island of Rhodes), beta thalassemia occurs with a frequency of up to 20% of all T., and a- and beta thalassemia are also significantly common. T. is also common in the countries of Southeast Asia. Abnormal genes in various combinations, including with structural hemoglobinopathies, lead to the occurrence of St. 60 different T shapes.
On the territory of the USSR, a detailed study of the spread of beta thalassemia was carried out in certain republics of Transcaucasia and Central Asia.
Family cases of beta thalassemia have also been described among the peoples of Dagestan, Kazakhstan, Moldova, the Mordovian Autonomous Soviet Socialist Republic, as well as in Penza and a number of other regions of the RSFSR located in the European part. Alpha thalassemia (in particular, hemoglobinopathy H) has been identified in Azerbaijani, Georgian, Armenian, Tajik, Uzbek and Russian families.
How does thalassemia manifest?
- First of all, thalassemia is characterized by a severe form of anemia - a decrease in the amount of hemoglobin and red blood cells and, consequently, a violation of oxygen transfer and respiration. Anemia causes weakness, dizziness, and shortness of breath.
- In addition, with thalassemia, the body is often overloaded with iron : after all, less hemoglobin is formed and iron atoms remain in free form in the blood. Iron accumulates in organs and tissues and prevents them from functioning normally. First of all, the liver, heart and endocrine glands suffer from this.
- Another dangerous consequence of thalassemia is an increase in the size of the spleen . It is in this organ that the breakdown of red blood cells occurs, which increases during illness. The larger the spleen, the greater the risk of rupture, which is life-threatening for patients.
- Also, with thalassemia, bones are often deformed . This occurs because, due to the reduced number of red blood cells in the bone marrow, the areas of hematopoiesis expand. Bone marrow cells can even extend beyond the bones, forming thickenings called pseudotumors. This leads not only to skeletal deformation, but also to other symptoms - after all, bones can touch nerves and blood vessels.
How iron affects well-being and health
Treatment
- In cases of severe forms, transfusion of whole blood or red blood cells is indicated. However, they provide only a temporary effect and can cause complications;
- Today, the most effective is considered to be the transfusion of thawed, washed or filtered red blood cells, which do not often provoke side effects along with long-term administration of iron chelates;
- Iron chelate must be injected for a long time (over several hours) under the skin. Therefore, special preparations have been created that can be attached to clothing. A patient suffering from a severe form of the disease must receive this drug for five days a week for the rest of his life. To prevent local tissue damage, injection sites may need to be changed occasionally. If hemolytic crises occur, then it is necessary to introduce small zones of glucocorticoids;
- If the size of the spleen is enlarged, it is removed. This operation should not be performed on children under 5 years of age. Normal age is 8–10 years. The positive effect can be observed during the first year after the operation, and then the condition may worsen again. The risk of infectious diseases increases;
- Today, bone marrow transplantation is considered the optimal method. This is the only radical method of treating the disease. However, it is quite difficult to find a donor;
- Patients should follow a diet that contains large amounts of tannin: tea, cocoa, soy, nuts. These products help reduce iron absorption.
There is also a need for symptomatic treatment:
- in order to improve liver function, hepoprotectors are prescribed for at least one month;
- vitamin prescription. C, which helps remove iron from the body.
Complications
If the form of the disease is severe, then complications often appear as a result of the deposition of hemosiderin in the tissues. Cirrhosis of the liver and diabetes mellitus often develop, as well as cardiosclerosis, which can cause the death of the child.
Hemoglobin mutations and disease severity
Hemoglobin consists of two types of chains - alpha and beta. According to the gene chain in which the mutation occurred, thalassemias are also divided into alpha and beta types. At the same time, the alpha chain of hemoglobin in humans is encoded by two pairs of genes, and the beta chain is encoded by only one. Depending on the number of mutated alleles, thalassemias are divided according to severity.
So, if only one copy of the alpha-globin gene stops working correctly, the disease is almost asymptomatic. Disruption of two copies leads to a mild form of alpha thalassemia, three - to clinically significant disorders (this form is called hemoglobinopathy H). If all four copies of the alpha globin gene do not work correctly, most often death occurs at the fetal stage. The development of beta thalassemia occurs in a similar way. If one of the two copies of the gene encodes functional beta globin, thalassemia is mild. If there are mutations in both copies of the gene that affect the production of beta globin, a severe form of the disease is observed - Cooley's thalassemia. But if disturbances occur simultaneously in both the alpha and beta chains, the formation of hemoglobin is compensated and the disease passes more mildly. The fact is that the negative effect of mutations in the genes of hemoglobin chains is not only a decrease in the amount of oxygen-transporting protein, but also a decrease in the balance between the two types of chains. Because of this, a breakdown in only the alpha-globin gene can be even more dangerous than in two types of genes at once. However, of course, such combinations are extremely rare.
Iron deficiency anemia: who is at risk and how to prevent the disease?
Clinical picture
Among beta thalassemias, depending on the wedge, the patterns are distinguished into major, intermediate, minor and minimal forms.
Beta thalassemia major (Cooley's anemia) is characterized by elevated levels of HbF (50% or more of total Hb) or HbA2. In some cases, HbA2 and HbF are simultaneously increased (in this regard, beta thalassemia major is divided into A2-, F- and A2F-thalassemia).
Most often, beta thalassemia major is detected between the ages of 2 and 8 years. Characterized by growth retardation, abdominal enlargement due to hepatosplenomegaly, cranial deformation (“tower” skull, “Mongoloid” face) as a result of changes in the face and cranial vault during the prenatal period caused by osteoporosis; icteric coloration of the skin and visible mucous membranes is noted; fever. A factor complicating the course of the disease is the development of secondary hypersplenism (see Spleen). In severe cases, myocardial dystrophy (see) with heart failure (see) and pericarditis (see) are observed. From the blood side, signs of severe hypochromic anemia are revealed (color index approx. 0.5, decrease in hemoglobin content to 20-50 g/l), which begins to appear at the end of the first year of life. The number of red blood cells is reduced to 1-2 million, their morphology is sharply expressed. changes (aniso- and poikilocytosis, polychromatophilia, hypochromia, fragmentation, schizocytosis, the presence of target-like erythrocytes, basophilic punctation, normoblastosis). Target-shaped erythrocytes, characteristic of all types of T., have increased osmotic resistance. There is usually a decrease in the average volume of red blood cells and the average hemoglobin content per red blood cell. In a scanning electron microscope, a large number of erythrocytes with a changed configuration are observed - drop- and bell-shaped, etc. Other blood sprouts are characterized by leukopenia with lymphocytosis, neutrophilic leukocytosis with a shift to the left during hemolytic crises, pancytopenia with hypersplenism. During hemolytic crises (see) and after splenectomy (see), the number of normoblasts in the blood increases sharply, and high reticulocytosis is detected, which is inadequate to the degree of hemolysis. The iron content in the blood plasma is increased (hypersideremia), and dipyrrole pigment is detected in the urine. Signs of hemolysis are an increase in the content of unconjugated bilirubin, as well as urobilinuria and an increase in the content of stercobilin in the feces.
Beta thalassemia major is characterized by complications - trophic ulcers (see), cirrhosis of the liver (see), cirrhosis of the pancreas (sometimes with symptoms of diabetes mellitus) due to the development of hemosiderosis in the internal organs (see), which becomes especially pronounced as a result repeated transfusions. Characterized by delayed puberty, signs of polyglandular insufficiency (see) and uric acid diathesis (see), patol. bone fractures. Resistance is reduced due to inferior function of T- and B-lymphocytes, and the development of pneumonia and sepsis is possible.
Beta thalassemia intermedia is characterized by a more benign course than beta thalassemia major. Signs of the disease appear at a later age and are less pronounced. Anemia is moderate (hemoglobin 70-100 g/l with a red blood cell count of 2,500,000-3,000,000). Expressed morphol. changes in red blood cells. Splenomegaly is in some cases combined with hepatomegaly. Changes in the cardiovascular system and growth retardation are not typical in this form of T. The appearance of the patients is almost unchanged. The main complications are hypersplenism and damage to the skeletal system. Genetically, this group of patients is diverse. This includes, in addition to heterozygous carriers of beta thalassemia, patients with certain homozygous forms, hemoglobinopathy H, as well as various compounds, including patients with hemoglobinosis SF (see Sickle cell anemia).
Beta thalassemia minor is characterized by mild hypochromic anemia and morphol. changes in red blood cells; osmotic resistance of red blood cells is usually increased, serum iron levels are normal or slightly elevated.
Minimal beta thalassemia (Silvestroni-Bianco syndrome) is asymptomatic and is discovered accidentally during a biochemical and genetic study of families with the beta thalassemia gene. The hemoglobin content and the number of red blood cells in such patients are normal. Sometimes there is a slight erythrocytosis against the background of characteristic morphol. changes in red blood cells. The osmotic resistance of erythrocytes is increased or normal; the average hemoglobin content per red blood cell may be reduced.
The degree of ineffectiveness of erythrocytopoiesis in all heterozygous forms of beta thalassemia (intermediate, minor and minimal) is usually small. With intercurrent diseases and pregnancy, the course of the disease can become significantly more complicated. Heterozygous forms of beta thalassemia are characterized by an increase in HbA2 content (on average 5.1%, but not more than 10%); Along with this, approximately 20-50% of gene carriers experience a slight (up to 10%) increase in HbF.
δβ-thalassemia is benign. In heterozygous carriers, δβ-thalassemia is characterized by normal HbA2 content and increased HbF content (up to 20%); in homozygous carriers, HbF can be up to 100% in the complete absence of other hemoglobin fractions - HbA and HbA2 (so-called δβ°-thalassemia). HbF is unevenly distributed in red blood cells.
With the so-called In silent variants of heterozygous thalassemia, no changes in hemoglobin fractions are noted, although the wedge, signs of the disease and morphol. red blood cell changes may be observed. Such heterozygous carriers are identified during examination of families, in which patients with T. of varying degrees of severity are found.
Of the alpha thalassemias, the wedge has signs of its heterozygous form - hemoglobinopathy H; it represents hron. moderate hemolytic anemia. Splenomegaly is observed in 80% of cases, changes in the skeletal system in approximately 1/3 of patients, enlarged liver in 70% of patients, hemosiderosis of internal organs is absent.
Serum iron levels are often reduced. The content of total hemoglobin varies from 50 to 130 g/l. A relatively large number of red blood cells is combined with a low hematocrit due to a decrease in the average cell volume. Quantitative erythrocyte indices are significantly reduced. Typical for T. morphol. changes in red blood cells. The degree of reticulocytosis varies. Most red blood cells contain characteristic HbH inclusions. Hemoglobin electrophoresis (pH 9.0) reveals a fast-moving fraction of HbH, migrating ahead of HbA. The HbH content at pH 6.5 ranges from 10 to 30%. Sometimes Hb Bart's is also detected (within 4-7%). HbH is unstable and functionally defective. Anemia in this form of T. is exacerbated by intercurrent infections and taking oxidizing drugs (sulfonamides, etc.).
Homozygous alpha thalassemia (hydrops fetalis with Hb Bart's) is manifested by fetal death. This disease was not discovered in the USSR.
Diagnosis
establish on the basis of a wedge, pictures, rentgenol. research (beta thalassemia major is characterized by small areas of osteoporosis along with areas of hypertrophy of the skull bones - the so-called brush or hedgehog symptom, as well as transverse striations of both tubular and flat bones) and the entire complex of hematol. and biochem. studies in combination with family genetic analysis - determination of total hemoglobin, the number of erythrocytes and reticulocytes, hematocrit index, calculation of quantitative erythrocyte indices, determination of the osmotic resistance of erythrocytes, their morphology in a blood smear, identification of erythrocyte inclusions with their supravital coloring, determination of alkali-resistant hemoglobin by biochemical and cytochemical methods, electrophoresis of hemoglobin on cellulose acetate film (pH 9.0 and 6.5) and in other media with subsequent quantitative determination of hemoglobin fractions, study of the biosynthesis of hemoglobin polypeptide chains in vitro using labeled radioactive amino acids.
Differential diagnosis is carried out with other forms of hereditary and acquired hemolytic anemia (microspherocytosis, structural hemoglobinopathies, etc.). Heterozygous hypochromic forms of T. most often should be differentiated from iron deficiency anemia (see), since both conditions are characterized by the same signs - hypochromia and microcytosis.
Inheritance and epidemiology
At the same time, only five percent of the world's population are carriers of mutations in the genes of hemoglobin chains, and of these, slightly less than two percent of people are susceptible to symptoms of the disease. It is also worth considering that residents of different regions experience thalassemia with different frequencies. The disease primarily affects residents of the Mediterranean region, as well as the countries of Central and South Asia. In Russia, there are relatively few carriers of mutations associated with thalassemia - about one percent (for the beta type).
Carriers of the mutation may not even be aware of their status - often damage to just one copy of the gene does not lead to severe symptoms. However, mutation carriers run the risk of passing the disease on to their children: if two carriers have a child, he can receive both copies of the mutated gene from his parents at once with a probability of about 25 percent.
In the previous section, we already discussed that mutations in two copies of genes lead to a more severe form of thalassemia. But if you calculate the probability of such an event, it turns out to be only about a hundredth of a percent.
Causes
The cause of the disease is a gene mutation of the gene that is responsible for the synthesis of globin chains. Such a gene can be inherited from one parent or two. The child’s body produces fewer hemoglobin chains or does not produce them at all. The production of other chains that make up globin does not end. As a result, unstable protein components are formed that destroy blood cells. Then anemia manifests itself and glands are deposited in the internal organs.
Diagnosis of thalassemia
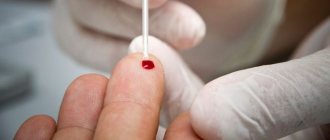
The most accurate way to diagnose thalassemia of any severity is to study the DNA structure. Establishing the DNA sequence will make it possible to detect disrupted gene variants and not only take timely measures in case of illness, but also plan pregnancy in the case of asymptomatic carriage.
Thalassemia can also be diagnosed using blood tests: general, biochemical, as well as determination of the hemoglobin fraction in the blood. These tests can determine the number of red blood cells, their volume, as well as the concentration of hemoglobin and the levels of some other substances in order to determine the type of thalassemia from the results obtained. If thalassemia is suspected, an abdominal ultrasound is also performed to assess the size of the spleen and liver, which are affected by the disease.
The Atlas genetic test helps identify pathogenic variants in the gene responsible for alpha thalassemia, detect risks associated with the manifestation of symptoms and transmission of the disease to children.
Therapy and prevention
To treat severe forms of thalassemia, blood transfusions are used to increase the number of red blood cells and restore oxygen transport, as well as chelation to remove the free form of iron that has not been incorporated into the “wrong” hemoglobin. In severe beta thalassemia, blood transfusions are needed frequently - every 3-4 weeks. However, now such therapy allows patients to lead a normal life and restores life expectancy. For mild forms of thalassemia, the indications are simpler - doctors advise maintaining a healthy lifestyle and moving more. Sports activities help maintain the mineralization of skeletal bones and prevent their deformation.
How effective is the treatment of beta thalassemia with Zinteglo?
The European regulator relied on the results of multiple phase I/II clinical trials of betibeglogen autotempsel: completed phase I/II HGB-205 (NCT02151526) and HGB-204 (Northstar, NCT01745120), ongoing HGB-207 (Northstar-2, NCT02906202) and HGB-212 (Northstar- 3, NCT03207009) phase III, long-term observational LTF-303 (NCT02633943).
The primary endpoint was the achievement of transfusion independence status: no need for blood transfusions over a period of 12 months or more while maintaining hemoglobin levels at an average of at least 90 g/L.
Data collected in mid-December 2021 for patients without the β0/β0 genotype showed that in HGB-205 and HGB-204, respectively, 75% of subjects (n=3/4) and 80% (n=8/10) reached Primary endpoint: transfusion independence lasted 21.2+ to 56.3+ months and mean hemoglobin level was 105.1 g/L (93–132).
For patients (n=3) who did not show adequate therapeutic efficacy of Zynteglo, a decrease in blood transfusion volumes by 100%, 86.9% and 26.8% and blood transfusion frequencies by 100%, 85.3% and 20 was demonstrated. 7% - during the period of 6–24 months of observation.
"Mosmedpreparaty"
Of HGB-207 participants without the β0/β0 genotype for whom data were collected for analysis, 80% (n=4/5) achieved the stated therapeutic status: transfusion independence lasted from 12.0+ to 18.2+ months with a mean hemoglobin level 124.2 g/l (115–126).
On a note
- Thalassemia is a severe hereditary disease that occurs due to mutations in the hemoglobin genes. It is characterized by weakness and dizziness, liver and heart disease, an increase in the size of the spleen, as well as bone deformation.
- There are two types of the disease - alpha and beta thalassemia. On average, in the world there are about five percent of carriers of mutations characteristic of both species, while in Russia it is only about one percent.
- Thalassemia is diagnosed using blood tests, ultrasound and genetic testing.
- To treat severe forms, frequent blood transfusions and removal of free forms of iron are used. For mild forms, maintaining a healthy lifestyle is sufficient.
Sources:
- Practical medicine, Hemoglobinopathies and thalassemic syndromes, 2015
- Subcellular Biochemistry, Hemoglobin: Structure, Function and Allostery, 2020
- Federal State Institution Federal Scientific and Clinical Center for Children's Health and Human Institutions of Roszdrav, Epidemiology of hemoglobinopathies in Moscow, 2008
- American Family Physician, Alpha and Beta Thalassemia, 2009
- NCBI, Thalassemia, 2021