Статья на конкурс «био/мол/текст»: «Мальчик, который остался без кожи». Это не название очередного фильма ужасов. Это документальный фильм режиссера Патрика Коллертона, запечатлевшего последние месяцы жизни 36-летнего Джонни Кеннеди, страдавшего редким генетическим заболеванием — дистрофическим буллезным эпидермолизом. Фильм привлек внимание пяти миллионов зрителей по всей Великобритании и помог собрать 500 000 фунтов стерлингов на благотворительность для международной ассоциации DEBRA, чья деятельность направлена на изучение и лечение буллезного эпидермолиза. Жизнь людей с этим диагнозом и их родных — это путь, который осилит не каждый: он тяжел как физически, так и морально, и требует мужества и невероятной воли к жизни. Детей, страдающих этим недугом, называют детьми-бабочками, сравнивая хрупкость их кожи с хрупкостью крыла бабочки. Что же такое буллезный эпидермолиз и каковы прогнозы на будущее?
Обратите внимание!
Эта работа опубликована в номинации «лучшая обзорная статья» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Я знаю только один способ быть в ладу с собственной совестью: этот способ — не уклоняться от страдания.
Антуан де Сент-Экзюпери
Общие сведения
Буллезный эпидермолиз (сокр. БЭ) – это понятие объединяющее целую группу редких наследственных болезней, имеющих генетическую и клиническую гетерогенность. Чаще всего патологические процессы происходят в кератиноцитах и приводят к образованию пузырей с доброкачественным течением, возникновению эрозивных участков кожи и слизистых оболочек, повышенной ранимости и чувствительности кожных покровов к любым незначительным механическим травмам, развитию «механобуллезной болезни», которая еще называется синдром Бабочки. Встречаемость разных видов пузырчатки – всего 1 случай на 30-100 тыс. человек.
Ввел понятие буллезного эпидермолиза немецкий дерматолог Генрих Кёбнер еще в 1886 г., хотя подобные случаи кожных заболеваний встречались и ранее.
Продолжительность жизни при буллезном эпидермолизе
Несмотря на стремительное развитие всемирной медицины продолжительность жизни у пациентов с врожденным эпидермолизом – «детей-бабочек» чаще всего не высокая. Все зависит от подтипа заболевания, глубины и обширности патологических поражений. Простой буллезный эмидермолиз при правильном уходе и комплексной терапии протекает легче, с возрастом минимизируется количество пузырей и прогноз для таких пациентов благоприятный. Другие типы генодерматозной болезни, особенно при присоединении инфекций, чаще всего стафилококковой или стрептококковой, при развитии системных осложнений и сепсиса чаще всего приводят к летальному исходу в ранние годы жизни детей.
Лечение
К сожалению, в настоящее время БЭ неизлечим. Основная терапия направлена на предотвращение образования новых пузырей и эрозий, лечение ран и предотвращение их инфицирования. Главные задачи лечения — защита хрупкой кожи пациентов от механических воздействий (использование специальных повязок) и обработка уже существующих ран (применение антисептиков, наложение повязок).
Но наука не стоит на месте, и ученые всего мира отчаянно ищут лекарство от страшного недуга. Наиболее проработаны три перспективных вида терапии БЭ: протеиновая, клеточная и генная. Рассмотрим эти подходы подробнее.
Протеиновая терапия
При этом подходе в организм пациента вводится достаточное количество нормального белка. Исследования показали, что внутридермальное введение мышам с рецессивным дистрофическим БЭ очищенного человеческого коллагена VII приводит к формированию нормальных коллагеновых фибрилл в зоне соединения эпидермиса с дермой. При внутривенном введении очищенного белка наблюдается системное отложение коллагена VII в коже. Также было показано, что подобная терапия не только улучшает функциональное состояние кожи, но и способствует заживлению ран. Основываясь на результатах доклинических исследований, компания Lotus Tissue Repair, впоследствии вошедшая в состав Shire Pharmaceuticals, начала проведение расширенных доклинических исследований внутридермальных и внутривенных инъекций коллагена VII на модели рецессивного дистрофического БЭ у собак, а также первую стадию клинических исследований [3, 5].
Клеточная терапия
При этом способе лечения в организм пациента вводится необходимое и достаточное количество клеток, содержащих нормальный ген, кодирующий нужный белок.
Одним из перспективных видов клеточной терапии является внутридермальная инъекция аллогенных (полученных от здоровых доноров) фибробластов. Так, было показано, что подобный подход восстанавливает синтез коллагена VII и стабилизирует соединение эпидермиса с дермой в мышиной модели рецессивного дистрофического БЭ. Следующий шаг на пути к реализации этого вида терапии уже предпринят: нескольким пациентам с рецессивным дистрофическим БЭ были введены аллогенные фибробласты. И хотя спустя две недели после инъекции введенные клетки не обнаруживались, уровень продукции коллагена оставался повышенным как через две недели, так и через три месяца. Ученые считают, что основной терапевтический эффект инъекции аллогенных фибробластов заключается в увеличении продукции эпидермального фактора роста HB-EGF, а также в увеличении экспрессии гена коллагена VII в кератиноцитах и фибробластах реципиента. Однако механизм этого явления пока не установлен [3, 6].
Аллогенная трансплантация костного мозга также может быть перспективным способом лечения БЭ. Так, на мышиной модели рецессивного дистрофического БЭ было показано, что трансплантация костного мозга приводит к облегчению симптомов болезни. Вслед за обнадеживающими результатами были начаты клинические испытания подобного метода терапии. Однако пока результаты неутешительны: пять из двадцати пациентов умерли от прогрессии заболевания или осложнений после трансплантации [3, 6].
Генная терапия
Наконец, последний и, пожалуй, самый перспективный подход — генная терапия (рис. 6). В этом случае пациентам пересаживаются аутологичные (сделанные из собственных клеток) трансплантаты, в клетках которых с помощью методов генетического редактирования дефектный ген заменяется нормальным. И хотя подобная процедура не приводит к полному исцелению, она способна временно устранить симптомы БЭ, а также является относительно простой в техническом отношении.
Рисунок 6. Редактирование собственной ДНК в лечении БЭ. а — Принцип генной терапии. На первом этапе культивируют эпидермальные стволовые клетки из биоптатов кожи пациента с БЭ. Затем стволовые клетки подвергают процедуре генетического редактирования, в результате которой дефектный ген замещается нормальным. После того как пласт из генетически отредактированных кератиноцитов будет проверен на безопасность, его трансплантируют пациентам. б — Трансплантаты, сформировавшиеся из «правильных» кератиноцитов пациента с рецессивным дистрофическим БЭ. Рисунок из [7].
Первые успехи генной терапии были достигнуты в Италии в 2006 году, когда в эпидермальные стволовые клетки пациента с пограничным БЭ с помощью ретровирусной конструкции был доставлен нормальный ген LAMB3, кодирующий одну из субъединиц белка ламинина-332. Из «исправленных» клеток были созданы эпидермальные трансплантаты, которые затем были пересажены на ноги пациента. В результате из них образовалась полноценная в функциональном отношении кожа. Исследование показало, что выживание даже небольшого числа стволовых клеток в подобных трансплантатах приводит к успешному восстановлению нормальных функций кожи. Спустя восемь лет продукция целевого белка LAMB3 в коже пациентов всё еще сохранялась. Не было обнаружено ни пузырей, ни признаков воспаления, опухолевого роста или какого-либо иммунного ответа в области трансплантата. В июле 2014 года в Австрии провели вторую подобную операцию. Профессор Альфред Лэйн из Стэнфордского университета (Калифорния) вместе с сотрудниками начал клинические испытания подобной технологии, нацеленной на восстановление синтеза коллагена VII в эпидермальных стволовых клетках пациента с рецессивным дистрофическим БЭ. Спустя 30 дней после операции в областях трансплантации не было отмечено никаких отклонений, а уровень продукции коллагена VII был гораздо выше исходного [7].
Конечно, и в случае генной терапии существует множество сложностей. Так, замена гена с использованием вирусных конструкций и невирусных систем редактирования ДНК (ZFN, TALEN и CRISPR/Cas9)* связана с риском встраивания гена не в то место генома, которое являлось мишенью. Оценить последствия такого встраивания довольно сложно. Поэтому такой подход требует большой осторожности.
* — Методы ZFN, TALEN и CRISPR/Cas9 основаны на сайт-специфическом действии нуклеаз in vivo. Эти системы обычно состоят из двух модулей, один из которых распознает целевую олигонуклеотидную последовательность, а другой режет цепи ДНК: «А не замахнуться ли нам на… изменение генома?» [8], «CRISPR-системы: иммунизация прокариот» [9], «Мутагенная цепная реакция: редактирование геномов на грани фантастики» [10]. — Ред.
Другая проблема подобной терапии — возможный риск развития иммунного ответа на экспрессируемый новый белок. Особенно велик риск для тех пациентов, чьи клетки несли мутации, полностью исключающие синтез определенного белка. В то же время известно, что у некоторых пациентов с БЭ есть участки кожи, на которых не образуются пузыри и продуцируется повышенный уровень белка. Это явление названо обратным мозаицизмом, или естественной генной терапией, и может предотвращать развитие иммунной реакции на синтезируемый в результате генной терапии белок. Более того, такие «самоизлечившиеся» клетки можно использовать для клеточной терапии, и тогда потребность в генной коррекции исчезнет. Однако до сих пор попытки создать трансплантаты на основе таких клеток у больных с пограничным БЭ не увенчались успехом из-за малого числа подобных клеток в пересаживаемом материале [11].
Патогенез
Патология выявляется с рождения – на коже имеются пузырчатые образования с серологическим содержимым, которые при вскрытии примерно через 2-3 дня оставляют долго незаживающие эрозии, чаще всего приводящие к развитию атрофичной рубцовой ткани, гипер- либо гипокератоза, повторному развитию пузырей. Известны случаи манифестации болезни и в более позднем возрасте – в младенчестве, раннем детстве и даже юношестве.
Поражение кожи — возникновение язв и пузырей вызывают любые механические воздействия, поэтому больше всего страдают участки с наиболее частым трением – подмышки и различные складки, места прилегания одежды и лямок.
В основе патологии лежат мутации генов, кодирующих белки различных слоев кожи – базальных мембран, дермы. При этом происходит нарушение баланса в системе ферментов и ингибиторов, белки становятся объектом атаки защитных систем организма – ферментативного цитолиза, вызывающего отек цитоплазмы, разрыв клеточных оболочек и как следствие — нарушений межклеточных связей, структуры эпидермиса с образованием внутри- либо субэпидермальных булл и везикул.
Новая кожа для «бабочки»
В ноябре журнал Nature
опубликовал статью, авторы которой, биологи и врачи из Италии и Германии под руководством Микеле де Лука, рассказали об удачном опыте по пересадке трансгенной кожи пациенту, страдающему тяжелым генетическим заболеванием — буллезным эпидермолизом. Благодаря тому, что новую кожу вырастили на основе собственной кожи пациента, она прижилась и, главное, позволяет пациенту — в настоящее время девятилетнему мальчику — чувствовать себя здоровым. Это событие — настоящий прорыв в области лечения так называемых «детей-бабочек». О его значении и клинических перспективах, а также о том, что такое буллезный эпидермолиз вообще, каковы его виды и особенности, сколько пациентов страдают от него в России и какую помощь им оказывают, мы поговорили с сотрудниками благотворительного фонда «Дети-бабочки».
Буллезный эпидермолиз (БЭ) — редкое генетическое заболевание, при котором на коже и слизистых оболочках возникают пузыри и эрозии. Оно может затрагивать какие-то определенные участки кожи (чаще всего — стопы и кисти), а может поражать и всю ее поверхность. Болезнь протекает иногда очень тяжело — кожа у заболевших чувствительна даже к незначительным механическим воздействиям, при некоторых формах БЭ на ней очень легко появляются повреждения.
Больным буллезным эпидермолизом требуются постоянный уход и лечение, направленные на предотвращение возникновения травм и устранение зуда, боли и других осложнений. Болезнь сказывается на многих внутренних системах организма. Поэтому пациенты нуждаются в мультидисциплинарном подходе лечения: в активном динамическом наблюдении и лечении не только у врачей-дерматологов, но и у других специалистов, таких как хирурги, стоматологи, гастроэнтерологи, педиатры, онкологи, офтальмологи, гематологи, психологи, диетологи.
Что происходит с кожей
БЭ проявляется в раннем детстве и может существенно влиять на качество жизни, а также быть причиной преждевременной смерти. Детей, страдающих от этого заболевания, часто называют «бабочками» — по аналогии с хрупкими насекомыми, пыльца на крыльях которых не выдерживает даже самого легкого прикосновения.
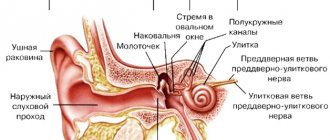
Напомним, что кожа человека состоит из эпидермиса, дермы и подкожно-жировой клетчатки. Эпидермис, в свою очередь, состоит из пяти слоев, самый нижний из которых располагается на базальной мембране. Базальная мембрана с верхней стороны называется светлой пластинкой кожи (lamina lucida), а со стороны дермы — плотной пластинкой (lamina densa). Белки базальной мембраны обеспечивают «заякоривание» эпидермиса в дерме.
БЭ возникает из-за мутаций в генах, кодирующих белки различных слоев кожи. В зависимости от того, в каком именно слое образуются пузыри, БЭ подразделяют на четыре типа. У простой формы
мишенью являются белки верхних слоев эпидермиса, кератины, плакофилин-1, десмоплактин и другие.
Пограничная форма
затрагивает уровень светлой пластинки кожи — а именно, белки ламинины, коллагены XVII типа и интегрины. При
дистрофическом БЭ
нарушены коллагены VII типа из сосочкового слоя дермы, ниже плотной пластинки кожи. Наконец, если пузыри образуются на разных уровнях, это называется
синдромом Киндлера
и связано с мутациями в гене белка киндлина-1.
Разные белки выполняют разные функции, но в целом все это — структурные белки, которые обеспечивают целостность слоев кожи и их связь друг с другом. Когда этих белков нет или их структура нарушена, кожа становится очень уязвимой для повреждаться, а кроме того, в ней постоянно образуются пузыри, похожие на ожоги. Один из наиболее распространенных и неприятных симптомов заболевания — частая боль.
Благотворительный фонд «Дети-бабочки»
Поделиться
Откуда появляется болезнь
«Сломанные» гены наследуются от родителей, по аутосомно-доминантному или аутосомно-рецессивному типу. «Аутосомное» наследование означает, что поломки происходят в генах, расположенных не в половых хромосомах (X или Y), — то есть с полом эта болезнь напрямую не связана. В случае «доминантного» наследования она проявляется даже тогда, когда сломан всего один из родительских генов, а в случае «рецессивного» (более распространенного) наследования необходимо, чтобы ребенку достались мутации от обоих родителей.
При пограничном БЭ мутации чаще всего обнаруживаются в гене LAMB3. Обычно пограничный БЭ наследуется по аутосомно-рецессивному типу, однако известны, например, случаи соматического мозаицизма, когда в каких-то клетках дефект проявляется, а в каких-то — нет, и тогда кожа начинает напоминать мозаику, то есть становится неоднородной с точки зрения заболевания.
Как ставят диагноз и лечат
Диагноз «буллезный эпидермолиз» врачи ставят без затруднений, чаще всего сразу понятна и конкретная форма заболевания. Более детальная диагностика может проводиться с помощью исследования образцов кожи, взятых при биопсии, методом трансмиссионной электронной микроскопии или иммуногистохимии (непрямой иммунофлюоресценции). При этом определяется отсутствие или снижение уровня разных структурных белков клеток эпидермиса (кератиноцитов) и базальной мембраны. Таким образом БЭ классифицируют — выявляют структурный белок, ген которого у пациента сломан, и получают клинический прогноз. Также проводят и генетические анализы, достоверно подтверждающие диагноз.
До сегодняшнего дня ни для одной из форм БЭ не существовало методов лечения, за исключением симптоматических — наложения повязок и обработки ран. Пациенту нужно постоянно прикладывать усилия к тому, чтобы его кожа как можно меньше травмировалась — помимо сильных неудобств и болей, кожа при БЭ подвержена инфекциям, а эрозии (разрушенные участки кожи, возникающие на месте поврежденных пузырей) существенно повышают риски развития онкологических заболеваний.
Трансгенная кожа
Недавно в научном и медицинском сообществах произошел большой прорыв — итальянские и немецкие ученые под руководством профессора Микеле де Лука из Центра восстановительной медицины «Стефано Феррари» сумели создать трансгенную кожу из собственных клеток семилетнего мальчика с пограничным БЭ и заменить ею 80 процентов его кожи.
С помощью методов генной инженерии в клетки была внесена работающая копия гена LAMB3, в результате чего симптомы болезни в трансгенной ткани исчезли. За счет того, что работающая копия вносится и в стволовые клетки, такая ткань обновляет сама себя. С момента трансплантации прошло два года, и мальчик жив, кожа его здорова, он учится в школе и занимается спортом, хотя до этого лежал в больничной палате, ежедневно нуждался в морфине и питался только через трубку. Подробнее об этом можно почитать здесь.
В своей работе ученые также рассказывают о предыдущем своем пациенте, мальчике, которому в 2006 году пересадили кожу ног. С тех пор прошло 11 лет, и трансгенные участки до сих пор поддерживают свои нормальные функции. Это, в свою очередь, позволяет надеяться на успешные долгосрочные результаты такого рода операций.
Благотворительный фонд «Дети-бабочки»
Поделиться
Благотворительный фонд «Дети-бабочки»
Поделиться
С вопросами о буллезном эпидермолизе, о том, как врачи оценивают достижение Микеле де Лука, и о перспективах лечения БЭ в России мы обратились к врачу-дерматовенерологу Наталии Михайловне Марычевой, сотруднице благотворительного фонда «Дети-бабочки» и Московского научно-практического центра дерматовенерологии и косметологии.
N + 1: Насколько распространено это заболевание? Сколько людей болеют им в России?
Наталия Марычева:
В настоящее время нет точной статистики по болеющим буллезным эпидермолизом у нас в стране. В нашем фонде зарегистрировано около трехсот детей — это пациенты в возрасте до 18 лет, но это даже не все больные, обращающиеся к нам, — некоторые не регистрируются в фонде, так как не хотят афишировать болезнь ребенка, и мы им, конечно, тоже не отказываем в помощи: консультируем, делаем назначения. Понятно, что на самом деле в стране больных еще больше.
В научном сообществе считается, что разными формами БЭ болеет приблизительно один человек на 30–100 тысяч; вероятно, в России эти цифры не отличаются.
Что такое фонд «Дети-бабочки»
Благотворительный фонд «Дети-бабочки» был образован в 2011 году для всесторонней поддержки детей с диагнозом буллезный эпидермолиз. Фонд существует на частные пожертвования и государственные гранты, его финансированию также помогают корпоративная поддержка, партнерские проекты с компаниями и акции. Главная задача фонда — создание в России системы оказания квалифицированной медицинской помощи больным БЭ. Для этого в первую очередь необходимо обучить специалистов. В настоящее время у медицинского сообщества в России слишком мало знаний об этом заболевании. Своевременно поставленный диагноз, правильный уход с самого рождения с использованием специальных перевязочных средств и кремов открывает «ребенку-бабочке» дверь в нормальную жизнь — с некоторыми оговорками.
Поэтому образовательная программа — одно из самых важных направлений деятельности фонда. В рамках программы мы направляем врачей на обучение в лучшие медцентры и клиники Европы, специализирующиеся на лечении больных БЭ. Также в рамках программы наши врачи и медсестры обучают региональных врачей.
Три года назад по инициативе нашего фонда на базе ведущего педиатрического учреждения страны в Москве — Национального медицинского исследовательского центра здоровья детей Минздрава России — открылось первое и пока единственное медицинское отделение для «детей-бабочек». Здесь подопечные фонда со всей России проходят комплексное обследование и необходимое симптоматическое лечение.
С НМИЦ здоровья детей мы работаем в сотрудничестве по отлаженной системе. Мы определяем очередность детей, которые ложатся на лечение. Благодаря патронажу мы знаем, в каком состоянии наши дети. В экстренных случаях и новорожденных мы госпитализируем в срочном порядке.
Для наших подопечных лечение в НМИЦ здоровья детей бесплатно. Мы госпитализируем детей со всей России и оплачиваем дорогу для ребенка и родителя, перевязочные средства на время лечения и процедуры, операции и обследования, которые не входят в ОМС. Мы полностью сопровождаем подопечных по госпитализации: сбор документов, необходимые справки, анализы, вызов в федеральный центр, такси забирает из аэропорта или вокзала, отвозит в больницу и также обратно.
Одновременно в НМИЦ здоровья детей находится в среднем четверо наших подопечных, но бывает и восемь-девять.
Полина Реутова директор по связям с общественностью фонда «Дети-бабочки»
Поделиться
Какие прогнозы бывают по срокам жизни пациентов?
Это зависит от формы болезни. Мы не очень любим озвучивать цифры, в частности, из-за самих пациентов. Люди иногда приходят и сразу говорят о том, что знают о плохих перспективах своего ребенка, поскольку слышали про вероятность онкологических заболеваний.
Я могу сказать, что при очень тяжелых формах речь может идти, например, о нескольких годах жизни или даже меньших сроках. Однако прогнозы далеко не всегда такие плохие, очень многое зависит от формы заболевания, от индивидуальных проявлений его у пациента и от ухода. Вчера, например, ко мне приходил мальчик с дистрофической формой, наш постоянных пациент. У него замечательный уход. На улице, если смотреть на него, когда он рядом с с друзьями, невозможно понять, кто именно из них болен. Вблизи, при детальном рассмотрении, конечно, видно, что у него ранки на коже, нет ногтей, слизистые затронуты — но он бегает, играет, учится.
Психологический момент очень важен, мы обычно говорим родителям, чтобы они не думали о своем ребенке как о больном, а думали о нем как о ребенке с особенностями кожи. Для него самого это тоже очень существенно. До взрослого возраста пациенты с не самыми тяжелыми проявлениями болезни тоже успешно дорастают, заводят собственных детей.
Какое на данный момент существует лечение этой болезни?
В основном это уход за кожей, наложение специальных повязок, обработка антисептиками, обезболивание., устранение зуда Когда нам звонят и сообщают, что родился или нашелся ребенок с БЭ, мы сразу выезжаем, чтобы осмотреть ребенка, оценить состояние его кожи и здоровья, обучить маму, как ухаживать за кожей, какими пользоваться перевязочными средствами, кремами, спреями.
Как работает фонд «Дети-бабочки»?
Когда рождается ребенок-бабочка, родители оказываются один на один с болезнью и множеством вопросов. Какими пользоваться медикаментами? Как накладывать повязки? Как купать ребенка? Где искать врачей? В нашем фонде работает медицинская служба патронажа. Врачи и медсестра фонда, которые на сегодняшний день являются ведущими специалистами по БЭ в России, навещают «детей-бабочек» дома. Чтобы осмотреть, оценить состояние здоровья и развития, назначить лечение. А также при первичном патронаже — обучить родителей и медперсонал, как ухаживать за кожей ребенка и как жить с диагнозом. Врачи выезжают незамедлительно к новорожденным «бабочкам», новым подопечным и в экстренных случаях. Медицинская команда фонда всегда на связи с родителями подопечных. О новорожденных «бабочках» мы узнаем от врачей или от родителей, которые обращаются в фонд с просьбой приехать, обучить, помочь. Иногда наши же подопечные сообщают, что в их городе появился «ребенок-бабочка». Тогда мы ищем ребенка — через врачей, местный Минздрав. Важно найти ребенка в первые дни жизни, чтобы сохранить его кожу, научить родителей, объяснить им, что жить с этой болезнью можно, чтобы они не отказались от ребенка, находясь в состоянии мощнейшего стресса. Важно в этот момент поддержать и ребенка, и маму.
Почти все перевязочные материалы, которые требуются «бабочкам», — импортного производства и очень дорого стоят. В среднем уход за ребенком-бабочкой требует от 50 до 150 тысяч рублей в месяц, в зависимости от возраста и тяжести заболевания. С такой финансовой нагрузкой семьи не способны справиться самостоятельно. Все они нуждаются в помощи. Мамы вынуждены не работать, чтобы ухаживать за больным ребенком. Для 300 детей, которые сейчас зарегистрированы у нас в фонде, только на перевязочные средства требуется 375-400 миллионов рублей в год. Конечно, это неподъемная сумма для нас. И мы не можем обеспечить всех подопечных медикаментами в полном объеме. При этом важно понимать, что в России, по неподтвержденным данным, больных БЭ примерно 2,5 тысячи.
Важно, чтобы обеспечение «бабочек» медикаментами взяло на себя государство. Мы ведем большую работу в этом направлении. Налаживаем отношения и сотрудничаем с региональными министерствами и департаментами здравоохранения. По нашей инициативе в настоящее время в трех регионах «детей-бабочек» обеспечивают медикаментами из местного бюджета. С 2021 года начнут обеспечивать еще в трех регионах. Это большая работа, которую мы вели на протяжении нескольких лет. Наши врачи расписывают количество и наименования медикаментов по каждому ребенку. Ведь в случае с БЭ все индивидуально: одному ребенку подходят одни перевязочные средства, другому — другие.
Полина Реутова директор по связям с общественностью фонда «Дети-бабочки»
Поделиться
И никаких других способов лечения не существует?
Лечение на данный момент есть только такое, симптоматическое. Но вот эта новость о трансгенной коже, которая только что появилась, наделала очень много шума. Так что теперь можно надеяться на эту методику.
Я советовалась со своими коллегами, и мы понимаем, что процедура с этим мальчиком, которому пересадили почти всю кожу, — это был шаг, на который пошли люди, у которых других шансов не осталось. Но результаты очень впечатляют, а ведь это была далеко не самая легкая форма БЭ. Значит, и для других пациентов появилась надежда.
Кроме того, от пограничной формы до дистрофической, самой тяжелой, тоже в принципе, не так далеко с точки зрения методики трансплантации.
Учитывая, что БЭ — болезнь генетическая, можно ли предсказывать ее с достаточной степенью вероятности?
Да, и это делается в России. Болезнь можно определить на ранней стадии беременности. Не так давно мы помогли молодой женщине, у которой дистрофический БЭ, родить здорового ребенка. В последнее время эта практика становится всё более востребованной — в семьях, в которых есть случаи БЭ.
Разновидности болезни, насколько я понимаю, моногенные, то есть
обусловленные мутациями в одном гене.Поэтому и генетические тесты, видимо, не очень сложные?
Да, но разные мутации вносят разный вклад в нарушение работы каждого гена. Поэтому анализ генетических тестов — не такой однозначный и простой процесс как кажется. Тем не менее, предсказать можно многое.
То есть можно протестировать родителей и понять, с какой вероятностью могут заболеть их дети?
Да. Чаще всего такие тесты делают, когда в семье уже есть случаи буллезного эпидермолиза. Здоровые люди, по крайней мере у нас в стране, такие тесты сейчас не проходят. Кроме того, генетические тесты делают больным, чтобы уточнить диагноз, а также для сбора данных для статистики и изучения болезни. Ну и иногда чтобы что-то про сестер и братьев понять.
Бывает такое, что болезнь проявляется не сразу?
Очень редко. Обычно у родившегося ребенка сразу видны повреждения на коже — пузыри и эрозия. Раньше у врачей в нашей стране было много проблем с диагнозом, ведь легко подумать, что это просто какая-то кожная инфекция, если вообще не знать о таком явлении, как БЭ. Ведь это достаточно редкое заболевание, и многие дерматологи просто не могли его распознать. За последние несколько лет ситуация изменилась, во многом благодаря работе нашего фонда. Например, родители или врачи обращаются к нам, и мы помогаем, в том числе, поставить диагноз.
Как вы считаете, методика пересадки трансгенной кожи — это настоящее решение проблемы?
Нет, потому что существует много разных форм БЭ и их нюансов. Даже при одной и той же форме эпидермолиза у разных детей он может выражаться в совершенно разных симптомах. Правильно будет сказать, наверное, что это лечение точно подходит для некоторых форм эпидермолиза.
Кроме того, буллезный эпидермолиз часто затрагивает, например, слизистые оболочки дыхательных и пищеварительных путей, в том числе во рту, и это тоже необходимо лечить. В исследовании Де Лука об этом ничего не говорится, лечение сфокусировано именно на эпидермисе. Но и это уже очень и очень много.
С одной стороны, можно сказать, что это все равно симптоматическое лечение, ведь само генетическое заболевание у ребенка после пересадки кожи никуда не делось. Но с другой стороны, ученые говорят, что теперь ему не требуется никаких повязок, никакого постоянного лечения и ежедневной обработки кожи, которая другим детям с таким диагнозом необходима не только для того, чтобы на улицу выйти, — чтобы просто сесть в кровати.
Много разговоров идет о риске перерождения трансгенных стволовых клеток в раковые. Как вы оцениваете эти перспективы? Насколько это действительно опасно и стоит ли говорить об этом здесь?
При наличии у человека буллезного эпидермолиза риски онкологических заболеваний очень высоки. Когда кожа постоянно повреждается, возникает ее эрозия и в этих участках очень часто появляются раковые образования. Так что эта болезнь сама по себе опасна именно с онкологической точки зрения.
Но в случае с трансгенной кожей ученые говорят, что встраивание новых генов происходит в преимущественно некодирующие участки генома, так что риски не так велики, как могли бы быть. Кроме того, это несопоставимые вещи — тяжелая форма болезни, при которой человека надо как можно скорее спасать, или небольшой потенциальный риск развития рака в будущем.
Пока что у речь идет всего о двух пациентах, тогда как для полной картины нужно гораздо больше наблюдений, но в целом это совершенно разные категории риска.
Что значит жить с диагнозом БЭ?
Буллезным эпидермолизом в России начали заниматься в 2011 году, когда был образован наш фонд. И заболевание внесли в список орфанных только в 2011 году, по нашей инициативе. Семьи с детьми-бабочками жили в некоем параллельном мире: медикаментов нет, господдержки нет, специалистов, которые могут помочь, нет, а больные есть. Выживали сами по себе, как могли. Поэтому статистика взрослых «бабочек» с тяжелой формой БЭ плохая, их крайне мало. Но в других странах, где заболеванием занимаются намного дольше, например в Англии, США (там больных БЭ намного больше, чем в России) статистика хорошая. Недавно мы вместе с издательством ЭКСМО выпустили художественную книгу «Моя жизнь в его лапах», она продается в книжных магазинах. Это реальная история женщины из Англии по имени Венди Хиллинг, у которой дистрофический БЭ. Венди сейчас 68 лет. Она родила и воспитала двух здоровых детей. Замужем, получила образование, работала. Конечно, там описаны и очень тяжелые моменты, связанные именно с БЭ. У Венди серьезные проблемы со слизистой дыхательных путей, ей нельзя плакать, сильно смеяться, иначе она может задохнуться. Но это вдохновляющая история.
Весной 2021 года одна наша взрослая «бабочка» родила здоровую дочку. Во время беременности мы провели пренатальную диагностику, именно на БЭ. Анализ показал, что плоду передалась болезнь. Тогда мама приняла решение прервать беременность. А вот при второй попытке обследование показало, что плод здоров. И она, спокойная и счастливая, проходила всю беременность, зная, что ребенок будет здоровым. Дочке сейчас уже полтора года.
Те дети, которых мы ведем с самого рождения, по состоянию кожи и здоровья сейчас намного лучше, чем те, которых мы находим, когда им уже пять, семь и больше лет.
Полина Реутова директор по связям с общественностью фонда «Дети-бабочки»
Поделиться
Когда новая методика может дойти до широкого применения в медицине?
Сложно сказать точно, потому что впереди еще много клинических испытаний. Наверное, пять–десять лет, скорее даже пять. Другой вопрос, когда эта методика придет в Россию.
Сейчас многих детей отправляют лечиться за границу, в частности, на деньги благотворительных фондов. А могут ли «дети-бабочки» вообще куда-то летать?
Конечно, ребенок с БЭ может вылететь за границу для получения лечения. И лет пять-семь назад им много приходилось это делать, даже для самых простых медицинских процедур, вроде прививок, постановок пломб и так далее. Сейчас ситуация изменилась, появилось необходимое оборудование, врачи с клиническим опытом, и все эти рутинные операции теперь делаются на территории России. Даже проводится баллонная дилатация, обследование со стороны слизистой пищеварительной системы, лечение зубов, в том числе под наркозом, операция по разделению пальцев. Теперь, чтобы расширить пищевод, пациентам с БЭ уже не приходится летать за границу.
Но в случае с пересадкой трансгенной кожи, поскольку речь идет о совершенно беспрецедентной методике, трудно говорить, как именно все будет устроено. Наш фонд является членом ассоциации DEBRA International. Это международное сообщество, занимающееся проблемой БЭ, там врачи, клиницисты, ученые, которые проводят исследования, участвуют в конференциях и постоянно следят за происходящим. Как только метод себя полностью оправдает, а лечение будет безопасно и проверено, мы об этом узнаем: куда ехать, как это все будет проходить и сколько стоить. Наши подопечные, наравне с детьми других стран-членов DEBRA, будут иметь возможность получить этого лечение. Сейчас необходимо провести молекулярное исследование у наших подопечных, чтобы у каждого определить ген, в котором произошла ошибка. И мы планируем это делать.
Материал подготовила Анна Казнадзей
Классификация
Изучая на ультраструктурном уровне морфологию кожи (количество, виды таких структур кожи как нити кератина, десмосомы, гемидесмосомы, якорные нити, якорные волокна и пр.) и локализацию пузырчатых образований в слоях эпидермиса. Выделяют три основных типа механобуллезного эпидермолиза, которые подразделяются на более чем 30 подтипов в зависимости от фенотипических и генетических особенностей больных, а также типа наследования:
- Простой тип (эпидермолитический согласно классификации Пирсона 1962 г.) – наиболее распространненный, встречается в 75% случаев и характеризуется образованием пузырей в верхних слоях эпидермиса на коже стоп, кистей рук, в тяжелых случаях – всего тела. Разделен на 2 основных подтипа супрабазальный, при котором белками-мишенями становится плакофилин-1, десмоплактин и пр., а также базальный подтип, вызывающий изменения структуры альфа-6/бета-4 –интегрина, причем кожные нарушения могут быть локализованными, генерализованными (пузыри возникают группами) и в виде пятнистой пигментации. Наиболее часто встречающимися клиническими формами является летальный акантолитический, поверхностный, с мышечной дистрофией, с атрезией привратника, огнасский, мигрирующий кольцевидный и т.д.
- Пограничный тип (люцидолитический) ведет к формированию пузырей в области светлой пластинки базальной мембраны и встречается в форме подтипа Херлитца, нарушающего строение ламининов 332 и 5, а также других подтипов вовлекающих в патогенез не только ламинины, но и коллаген XVII типа и α альфа-6/бета-4 –интегрин. Кроме того, выделяют клиническую форму инверсионную, с поздним началом, в виде ларинго-онихо-кутанного синдрома.
- Дистрофический тип (дермолитический) – поражает верхнюю часть сосочкового слоя толщи дермы, который бывает доминантного и рецессивного подтипа и развивается за счет нарушения структуры коллагена четвертого типа. Он бывает генерализованным, периферическим, претибиальным, центрипетальным, пруригинозным, поражающий только ногти, инверсным.
- Отдельной формой, наиболее редкой и малоизученной считается синдром Киндлера, так как пузыри могут образовываться в разных слоях эпидермиса и вызвано это нарушением структуры белков – киндлинов-1. Они выявляются сразу при рождении ребенка, чаще всего на кожных покровах рук и ног. В последствие может наблюдаться возникновение дистрофических изменений ногтей, развитие кариеса, пародонтита, различных заболеваний ротовой полости, ЖКТ, оболочек глаз, мочеполовой системы. С возрастом количество новообразующихся пузырей сокращается, но кожа все равно остается тонкой, легко ранимой, чувствительной, капиллярная сетка располагается слишком близко к поверхности эпидермиса.
Клинические проявления БЭ
Буллёзный эпидермолиз проявляется уже во время родов: кожа ребёнка травмируется, когда он проходит через родовые пути. Обычно страдают нос, подбородок и пятки. В редких случаях болезнь дает о себе знать на 1-6 месяце жизни. С возрастом, в зависимости от типа БЭ, могут проявляться новые симптомы заболевания.
Основным клиническими проявлениями буллёзного эпидермолиза являются пузыри на коже, появляющиеся на местах трения, ушиба, давления, при повышении температуры тела, окружающей среды или спонтанно. Образование пузырей может возникать и на слизистых оболочках в любом органе. Чаще всего поражается слизистая оболочка полости рта, пищевода, кишечника, мочеполовой системы, слизистой глаз.
Причины буллезного эпидермолиза
Основные причины возникновения патологии — мутации более чем в 10 генах, отвечающих за кодирование белков различных слоев кожи, чаще всего в 75% — это гены KRT5 и 14, LAMC2, LAMA3, LAMB3, COL17A1. Как указывает онлайн-ресурс Википедия почти для каждого из установленных подтипов БЭ удалось выявить мутации в определённых генах, среди них чаще всего встречаются:
- миссенс точечные мутации;
- нонсенс точечные мутации;
- делеции и инсерции – потери и соответственно вставки участков хромосом;
- мутации сдвига рамки считывания;
- сплайсинг.
Тип наследования буллезного эпидермолиза встречается как аутосомно-рецессивный (наиболее часто), аутосомно-доминантный, так и однородительская дисомия, и соматический мозаицизм.
Факторы, провоцирующие врожденный буллезный эпидермолиз
Врожденный синдром Бабочки может развиться даже у здоровых родителей, не несущих мутированные гены. Спонтанные мутации возникают внутриутробно и причиной тому становится:
- вредные привычки беременной женщины – курение, злоупотребление алкоголем;
- хаотичный прием лекарственных препаратов и прочие тератогенные факторы.
Литература
- Boeira V., Souza E., Rocha B., Oliveira P., Oliveira M., Rêgo V., Follador V. (2013). Inherited epidermolysis bullosa: clinical and therapeutic aspects. An. Bras. Dermatol. 88 (2), 185–198;
- Fine J.D. (2010). Inherited epidermolysis bullosa. Orphanet. J. Rare Dis. 5, 12;
- Shinkuma S. (2015). Dystrophic epidermolysis bullosa: a review. Clin. Cosmet. Investig. Dermatol. 8, 275–284;
- Альбанова В.И., Чикин В.В., Епишев Р.В. (2014). К вопросу о диагностике врожденного буллезного эпидермолиза. Вестник дерматологии и венерологии. 3, 53–59;
- Bruckner-Tuderman L., McGrath J.A., Robinson E.C., Uitto J. (2013). Progress in epidermolysis bullosa research: summary of DEBRA International Research Conference 2012. J. Invest. Dermatol. 133, 2121–2126;
- Soro L., Bartus C., Purcell S. (2015). Recessive dystrophic epidermolysis bullosa. A Review of disease pathogenesis and update on future therapies. J. Clin. Aesthet. Dermatol. 8 (5), 41–46;
- Murauer E.M., Koller U., Pellegrini G., De Luca M., Bauer J.W. (2015). Advances in gene/cell therapy in epidermolysis bullosa. Keio J. Med. 64 (2), 21–25;
- А не замахнуться ли нам на… изменение генома?;
- CRISPR-системы: иммунизация прокариот;
- Мутагенная цепная реакция: редактирование геномов на грани фантастики;
- Kiritsi D., Garcia M., Brander R., Has C., Meijer R., Jose Escámez M. et al. (2014). Mechanisms of natural gene therapy in dystrophic epidermolysis bullosa. J. Invest. Dermatol. 134 (8), 2097–2104..
Симптомы
Симптоматика буллезного эпидермолиза связана с нарушением структуры кератиноцитов и проявляется в виде:
- жжения, боли в местах поражений и облегчения при опорожнении пузырей;
- зуда во время заживления вскрывшихся пузырей – отслоения подсохшей крышки везикулы, что в дальнейшем может вызвать шелушение и пигменатцию;
- развития участков кожи с эрозией;
- повышения чувствительности и ранимости кожных покровов к любым механическим повреждениям;
- спонтанного возникновения напряженных пузырей, содержащих прозрачную бесцветную жидкость или геморрагическое содержимое.
Патология обычно обнаруживается сразу при рождении, ведь тонкая и «ломкая» кожа легко травмируется даже от прохождения по родовым путям. При этом кожные поражения имеют разную степень тяжести и распространенности даже в пределах семьи. Множественные распространенные пузыри могут вызвать смерть новорожденного, особенно при присоединении вторичной инфекции. Обострения обычно припадают на летнее – теплое время года, с возрастом распространенность пузырей минимизируется.
Дополнительные симптомы простого БЭ
Простые эпидермолитические поражения обычно возникают на кистях рук и на стопах либо могут покрывать всё тело. Кроме того может наблюдаться:
- развитие ладонно-подошвенного гиперкератоза распространённого или сливного вида;
- дистрофические изменения ногтевых пластинок;
- стеноз гортани;
- милии;
- гипер- либо гипопигментация;
- задержка роста.
Заживление пузырей чаще всего происходит без рубцевания, иногда даже напоминает простой герпес, но при этом очень высока вероятность рецидивов.
Простой буллезный эпидермолиз
Особенности течения пограничного БЭ
Этот тип отличается развитием патогномоничного симптома – возникновения участков обильной грануляционной ткани на различных частях тела (как на фото буллезного эпидермолиза пограничного типа), например, локализованной симметрично вокруг рта, в средней части лица, вокруг носа, на верхней части спины, в подмышечных впадинах или ногтевых валиках. Патология может привести к ониходистрофии и даже полной утрате ногтевых пластинок, большому количеству милий, серьёзных рубцов на теле, в том числе рубцовой алопеции, корок на коже, язв в ротовой полости, гипоплазии эмали и тяжелому кариесу зубов. Среди возможных системных осложнений обычно выявляют полиэтиологическую анемию, задержку роста, эрозии, кожные везикулярные высыпания и отслоение, стриктуры в желудочно-кишечном тракте. Риск их развития повышается в зависимости от площади, степени деструктивности и глубины кожных поражений.
Пограничнй буллезный эпидермолиз
Пограничный буллезный эпидермолиз характеризуется крайне высокой смертностью преимущественно в первые годы жизни, вызванной прекращением прибавки в весе, сепсисом, пневмонией или обструкцией трахеи и гортани.
Внекожные проявления дистрофического БЭ
Помимо генерализованных кожных поражений — рецидивирующего образования пузырей, эрозий, милий, атрофического рубцевания и утраты ногтей «синдром Бабочки» вызывает:
- нарушения работы желудочно-кишечного тракта;
- поражение мочеполового тракта и внешних глазных оболочек;
- хроническую анемию;
- остеопороз;
- задержку роста;
- уплотнение, дистрофию и даже утрату ногтевых пластинок;
- контрактуры различных суставов конечностей – локтевых, коленных, лучезапястных, плюсне-предплюсневых, плюснефаланговых и т.д.;
- повышенный риск развития новообразований, к примеру, агрессивной плоскоклеточной карциномы.
Дистрофический буллезный эпидермолиз
Что это за болезнь
Врожденный буллезный эпидермолиз включает в себя целую группу заболеваний (более 30 форм), которые объединены одним названием. Общий признак этих болезней – появление на коже пузырей при малейшей механической травме. Данную патологию называют еще болезнью бабочки, но не от того, что вырастают крылья у больных детей, а от характерной для них чрезмерной хрупкости и уязвимости кожных покровов (у бабочки легко повредить крылья). В механизме возникновения пузырей лежит сдвиг между верхними и нижними слоями кожи. Назвать болезнь данным термином предложил немецкий врач – дерматолог Генрих Кёбнер. Группа данных заболеваний генетически детерминирована и может наследоваться по аутосомно-рециссивному и по аутосомно-доминантному типу, доказано, что мутации затрагивают более 10 генов.
Эпидемиология
Болезнь буллезный эпидермолиз распространена на территории 70 (всего 85) субъектов РФ. Частота ее составляет 0 – 19, 73 случаев на 1000000 населения. В России заболеваемость буллезным эпидермолизом достигает 1:50 тыс. – 1:300 тыс. населения.
Исследователи патологии выявили, что во многих странах мира в структуре болезни преобладает простой буллезный эпидермолиз, а в некоторых странах – дистрофический буллезный эпидермолиз. Половые различия для данной патологии не характерны. Отмечено, что среди зарегистрированных пациентов преобладают дети и подростки, что связано с высокой смертностью таких больных.
Анализы и диагностика
Наибольшее значение в постановке диагноза играет биопсия — исследование образцов кожи при помощи трансмиссионных электронных микроскопов, позволяющих осуществить визуализацию и провести полуколичественный анализ различных структур эпидермиса. Благодаря доступности моноклональных и поликлональных антител против белков разных слоев эпидермиса, участвующих в патогенезе буллезного эпидермолиза на сегодняшний день всю большую популярность завоёвывают иммуногистологические методы.
Иммуногистохимический метод исследования и метод непрямой иммунофлюорисценции позволяют определить состояние экспрессии структурных белков с наследственными дефектами в клетках кожи — кератиноцитах и базальных мембранах (основы эпителия, выполняющей барьерную и трофическую функцию), а также схему распределения протеидов в ранее образованных или искусственно спровоцированных пузырях, в том числе — их глубину локализации.
Благодаря современным методам ДНК-диагностики удается быстро классифицировать разновидность патологии, выявить структурные белки, подвергшиеся мутации и составить клинический прогноз. Инновационный метод генетического анализа – прямое секвенирование дает возможность выявить мутации, их тип и локализацию, достоверно подтвердить диагноз.
Помимо этого важную роль играет сбор семейного анамнеза и истории болезни пациента, комплексное обследование всего организма, проведение лабораторных анализов.
Обсуждение
Диагноз ВБЭ в России преимущественно устанавливают на основании анамнеза и клинической картины, как и в описанных нами случаях. Заболевание включено в перечень орфанных, а пациенты нуждаются в обеспечении перевязочными материалами, лекарственными препаратами, продуктами лечебного питания и создании специальных условий в быту, что требует высоких материальных затрат. Однако выполнение этих мероприятий из-за отсутствия статистического учета больных ВБЭ осложняет оказание адресной помощи. В то же время вспомогательные лабораторные методы диагностики — трансмиссионная электронная микроскопия и иммунофлюоресцентное антигенное картирование в большей части регионов отсутствуют, что затрудняет идентификацию типа ВБЭ и прогнозирование длительности жизни индивидуума, необходимой ему в будущем медицинской помощи специалистов различного профиля. Для оказания им регулярной и полноценной паллиативной помощи целесообразно в процессе стационарного лечения вести обучение матерей по уходу за такими больными. Полномасштабный реестр и диспансерное наблюдение, а также алгоритм их маршрутизации в регионе повысят качество жизни этих пациентов. Эти вопросы требуют решения.
Авторы заявляют об отсутствии конфликта интересов.
У детей
Детей с диагнозом буллезный эпидермолиз называют бабочками, потому что их кожа настолько ранимая, нежная и незащищенная как крылья этих насекомых.
Главным способом уберечь свое будущее чадо от этого диагноза является ответственное планирование семьи и генетическое обследования. Не стоит забывать о важности скрининга, способного выявить отклонения в развитии плода. Например, важно пройти пренатальную диагностику простой пузырчатки, которую можно обнаружить на II триместре беременности по высокому содержанию фетопротеина в сыворотке крови.
Продолжительность жизни больных
Сколько живут дети-бабочки? Ответить на данный вопрос однозначно затруднительно. Продолжительность жизни больных с буллезным эпидермолизом прямо пропорциональна форме заболевания, степени повреждения строения генов, глубины повреждения кожи и общего состояния ребенка. К сожалению, до зрелого возраста такие больные доживают редко, и в первую очередь это связано с уходом за ними, что требует множество усилий, терпения и мужества. Дети с тяжелыми генерализованными формами болезни живут всего несколько лет и погибают еще в дошкольном возрасте, например, синдром Герлитца или Аллопо-Сименса. Легкие формы, особенно простого БЭ могут со временем войти в стойкую ремиссию, а образование пузырей связано лишь с травматизацией кожи.
Диета при буллезном эпидермолизе
Диета 15 стол
- Эффективность: лечебный эффект через 2 недели
- Сроки: постоянно
- Стоимость продуктов: 1600-1800 рублей в неделю
Вне зависимости от возраста больного и типа патологии лечебная диета должна полностью компенсировать потери белков, солей, жидкости, которые вызваны патологическими изменениями внешних покровов и внутренних систем. Если речь идет о детях грудного возраста, то наиболее рекомендованным является материнское молоко, дополненное докормом белковой пищей – не менее 20%. Питание должно быть дробным, механически, термически и химически щадящее, есть следует маленькими порциями. С возрастом в рацион должны быть введены:
- некислые фруктовые соки и пюре;
- нерафинированное растительное масло добавлять в завтрак для облегчения дефекации и насыщения полезными жирами;
- овощи богатые растительной клетчаткой, например, капуста, кабачки, сухофрукты, свекла, которые лучше употреблять перетертыми и отварными.
Под строгий запрет попадает карамель и леденцы, сладости, печенье, спиртные напитки, острые и пряные блюда – любую еду, которая может спровоцировать образование буллезных поражений в пищеводе и других отделах ЖКТ.