Reasons for the development of pathology
There are many causes for the development of pulmonary fibrosis. These include:
- Diseases of the respiratory system (COPD, pneumonia, tuberculosis).
- Systemic connective tissue diseases (scleroderma, SLE, rheumatoid arthritis).
- Taking a number of medications (cytostatics, antiarrhythmic drugs).
- Vasculitis of various origins.
- Harmful production factors (asbestos, silicates, etc.).
Idiopathic pulmonary fibrosis is also isolated when the cause of the pathological process cannot be determined.
How to avoid complications
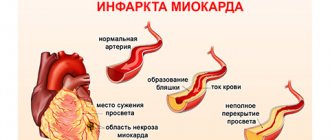
Why is pulmonary fibrosis dangerous after coronavirus? With a large area of fibrous growths, not only the lungs suffer. The appearance of foci of calcification in the lungs after coronavirus reduces the volume of inhaled and exhaled air. Complications arise in the functioning of the cardiovascular system - the heart, in order to saturate the organs with oxygen, begins to pump blood faster. Deformation of blood vessels appears, an increase in pressure is observed, and blood clots form.
The consequences from the nervous system are expressed by manifestations of oxygen starvation of the brain: a decrease in cognitive functions, performance, memory problems, rapid fatigue, and the development of a persistent depressive state.
Timely treatment of pulmonary fibrosis after coronavirus gives a favorable prognosis. Further rehabilitation is aimed at eliminating the pathological condition and minimizing the risk of complications.
Symptoms of pulmonary fibrosis
At the initial stage, the disease manifests itself as general weakness and increased fatigue. Patients experience pale skin with a bluish tint, especially in the nasolabial triangle, nose and limbs. Many patients report drowsiness during the day and insomnia at night, as well as unmotivated weight loss.
Then shortness of breath joins the above complaints. At the beginning, it bothers the patient only during physical activity, but as the pathology progresses, it begins to occur at rest.
Some patients have a dry or wet, unproductive cough. In the second case, a scant amount of viscous mucous sputum is detected. The appearance of purulent discharge indicates the addition of a secondary bacterial infection.
In patients suffering from pulmonary fibrosis for a long time, there is a change in the shape of the nails, which become like “watch glasses” and the terminal phalanges of the fingers like “drumsticks”.
At the peak of the disease, heart failure occurs, manifested by peripheral edema, palpitations, arrhythmia, pulsation of the neck veins, and severe shortness of breath at rest.
Idiopathic pulmonary fibrosis (IPF), which morphologically corresponds to common interstitial pneumonia (IP), can claim to be one of the most prognostically unfavorable variants of interstitial pulmonary lesions, which is characterized by a rapid rate of formation of practically irreversible PF with severe respiratory failure and pulmonary hypertension. The treatment tactics for IPF remain predominantly pathogenetic, not fundamentally different from those used for other IP; Moreover, its features are largely determined by the individual experience of a particular clinical center. In this regard, the importance of consensus recommendations of a number of professional societies published in 2011 can hardly be overestimated from the point of view of the high relevance of attempts to unify the tactics of managing IPF. The difficulty of implementing these efforts in practice is understandable, especially given the clear lack of clinical studies involving patients with IPF that would be conducted taking into account the standards of evidence-based medicine.
The 2011 guidelines defined IPF as a specific form of chronic progressive fibrosing PV of unknown origin, observed primarily among adults, especially the elderly, and without features of a systemic disease. The diagnosis of IPF requires the mandatory exclusion of the provoking role of known etiological factors of interstitial lung diseases (for example, occupational ones), and the presence of characteristic signs of ordinary IP, recorded with high-resolution computed tomography (CT). It is recognized that the accuracy of the diagnosis of IPF increases with a lung biopsy (preferably surgical, but not transbronchial), and the course of the disease, as a rule, is characterized by steady progression: only in a small part of patients the pulmonary process stabilizes, but repeated exacerbations are possible. It should be emphasized that IPF should certainly be considered as one of the most unfavorable variants of interstitial lung diseases: it is in patients suffering from it that the rate of formation of irreversible PF is maximum up to the stage of honeycomb lung and severe chronic respiratory failure. The mentioned features of the clinical evolution of IPF are largely related to the expression profile of inflammatory cytokines and chemokines in the lung tissue, typical for this disease: in comparison with other forms of PV, it is in IPF that the expression of transforming growth factor-β is maximum [3].
Features of the expression of mediators of inflammation and fibrosis, as well as existing descriptions of familial cases of IPF [4], make it necessary to discuss the contribution of genetic determinants to its development. At the same time, it is already obvious today that IPF is not a monogenic disease; it will not be possible to identify those genetic determinants that would clearly form a predisposition to the disease and/or modulate its development in a significant proportion of patients. Repeated attempts have been made to demonstrate the contribution of polymorphisms of individual genes to the progression of IPF. It has been demonstrated that in smokers suffering from IPF, the TT755 variant of the matrix metalloproteinase-1 gene, detected on fibroblasts, is significantly more common [5]. A recently published meta-analysis by NM Korthagen et al. (2012) [6] showed the significance of individual variants of the gene encoding the interleukin-1 receptor antagonist protein as a risk factor for IPF. An association of IPF with carriage of the 308A allele of the tumor necrosis factor α gene in both hetero- (the probability increases by 2.9 times) and homozygous (the probability increases by 13.9 times) form has also been shown [7]. G6A polymorphism of the angiotensinogen gene, as demonstrated by M. Molina-Molina et al. (2008) [8], may influence the course of IPF, in particular the severity of respiratory disorders, but not the formation of a predisposition to it. The intense progression of IPF may also be reflected by the maximum expression of matrix metalloproteinase-9 by cells from broncho-alveolar lavage [9]. Comparison of the genetic “portrait” of lung tissue obtained by biopsy in patients with IPF and pneumonitis caused by hypersensitivity reactions showed that IPF is characterized by predominant expression of genes for tissue remodeling, proliferation of epithelial cells and fibroblasts, while hypersensitivity is characterized by predominant expression of inflammatory genes and activation of T lymphocytes [10]. Overall, the 2011 guidelines emphasize that IPF should not be considered a genetically determined disease. At the same time, further search for certain genetic determinants of the development and progression of the disease is obviously justified, since it allows us to get closer to understanding the pathogenetic features of IPF, which in the future could become the target of specially developed targeted therapeutic strategies.
The 2011 guidelines discuss in detail the importance of other potential etiological factors for IPF, which are of interest primarily in the implementation of appropriate treatments. Among them, smoking is of particular importance. Back in the early 1990s. H. de Cremoux et al. (1990) [11] demonstrated that IPF patients who smoked were characterized by a predominance of lymphocytes in the broncho-alveolar lavage fluid, a worse response to glucocorticosteroids and worse survival. It has also been established that smokers suffering from IPF have the most severe respiratory disorders [12]. A multicenter clinical study that included 248 patients with IPF showed that smoking is associated with a significant 1.6-fold increase in the risk of developing IPF. Former smokers were also characterized by an increased risk of IPF (odds ratio – 1.9), with the highest risk in smokers with 21 to 40 pack years of experience (odds ratio – 2.3) [13]. It has also been established that smokers, including former smokers, who have IPF, have significantly worse survival [14]. It can be argued that all patients with IPF should use all possible means of treating nicotine dependence, although smoking is not an obligate risk factor for IPF.
Among the possible determinants of IPF, various environmental factors are also named, including metal dust (copper, steel, lead) [15]. Organic dust can also be considered as potential factors for the development of IPF: there are known cases of IPF development among agricultural workers, poultry farmers, and livestock farmers [16]. However, from an epidemiological point of view, the connection between IPF and environmental factors has not yet been convincingly confirmed, and each specific patient in whom such a connection is suspected needs a particularly detailed examination and discussion, obviously by a council of specialists. Interpretation of the role of exogenous factors in the development of IPF must be very cautious, especially when the patient's occupational history is used as part of the evidence.
Various infectious agents are also discussed as provocateurs for the development of IPF, primarily viruses: Epstein–Barr, cytomegalovirus, herpes viruses type 7 and 8 [17, 18]. At the same time, despite the fact that the DNA of these viruses could be detected, including in samples of lung tissue obtained by biopsy, the frequency of their detection in general does not differ from that in patients with interstitial pulmonary processes of a different nature. In this regard, the importance of the vast majority of viruses, including Epstein-Barr and representatives of the herpes group, which are credited with a role in the development of many variants of immunopathological lesions of internal organs, in particular systemic diseases, as risk factors for IPF should not be exaggerated, especially unreasonably carrying out antiviral therapy in this category of patients.
Chronic HCV infection is considered one of the most realistic risk factors for IPF [19], although the frequency of its detection in this category of patients, according to various clinical studies, varies significantly. In the first half of the 1990s. T. Ueda et al. (1992) [20] demonstrated that, in comparison with healthy representatives of the control group, the presence of anti-HCV antibodies in the blood serum of patients with IPF can be detected almost 8 times more often. A study of native Italians with IPF showed that the presence of antibodies to HCV could be detected in 13.3% of them, only in 0.3% of blood donors and in 6.1% of patients suffering from other interstitial lung diseases, but not IPF [21]. There are indications that HCV infection is not only more common among patients with IPF, but may also be considered a risk factor for its development. Y. Arase et al. (2008) [22] observed a group of 6150 patients with HCV infection and 2050 patients with HBV infection, the average follow-up duration was 8.0 ± 5.9 for HCV-infected and 6.3 ± 5.5 years for HBV-infected . During the observation period, IPF developed in 15 HCV-infected patients and in none of the HBV-infected patients. The total incidence of IPF in the presence of HCV infection was 0.3% by the 10th year of observation and 0.9% by the 20th year. IPF, therefore, developed significantly more often among HCV-infected people compared to HBV-infected people. Risk factors for IPF in persons with chronic HCV infection included age ≥ 55 years, heavy smoking, or the presence of liver cirrhosis. It should be emphasized that the development of IPF during HCV infection can be associated with cryoglobulinemia and typical damage to internal organs: there are reports of the effectiveness of antiviral therapy (interferon-alpha) in this situation [23]. Apparently, each case of IPF associated with HCV infection should, at least initially, be considered as its extrahepatic manifestation; However, in such patients, the effectiveness of antiviral therapy in terms of the effect on the pulmonary process has not been established and for them, as a rule, glucocorticosteroids and cytostatics have to be used.
Recommendations of the 2011 version consider gastroesophageal reflux (GER) as one of the priority risk factors for IPF. It is believed that microaspirations of aggressive gastric contents observed with it can cause combined chemical, immunological and bacterial damage to the lungs, resulting in the formation of IPF [24]. According to some data [25], GER can be detected in the vast majority of patients with IPF, and it is in them that it reaches its maximum severity. GER can be detected in 65% of patients with IPF who are candidates for lung transplantation. In general, today we can say that treatment of GER is justified for all patients with IPF, since, although not being the main therapeutic approach, it can still help improve the prognosis. JS Lee et al. (2011) [26] recently published the results of an analysis of the effect of GER and its therapy on the survival of patients with IPF. A total of 204 patients were analyzed, of which 34% had complaints characteristic of GER, 45% had a confirmed history of GER, and 47% were taking medications for GER. It turned out that taking medications used to treat GER is associated with an increase in survival of patients with IPF and a decrease in the area of the LF assessed on CT. It is obvious that examination aimed at identifying GER and its most active treatment are justified for all patients with IPF.
The 2011 guidelines clearly state the diagnostic criteria for IPF:
• absence of possible causes of IPF (exogenous factors, including occupational, systemic diseases, medications);
• the presence of signs of conventional PV on high-resolution computed tomograms (a sufficient sign for those who have not undergone a lung biopsy);
• the presence of signs of conventional IP on high-resolution CT and characteristic morphological features in lung tissue samples obtained through surgical biopsy.
It is fundamentally important that this version of the recommendations clearly postulates that IPF is not only a clinical diagnosis and its confirmation requires the mandatory use of CT, and, if possible, surgical (but not transbronchial) lung biopsy. It should, however, be emphasized that justification for the use of these methods is impossible without a thorough analysis of the details of the anamnesis of the pulmonary process and the correct interpretation of the data obtained during a general clinical examination. In general, IPF is often diagnosed late, and the prescription of pathogenetic therapy is often preceded by several (at least 1.5) years of repeated courses of various antibacterial drugs [3], used for imaginary “chronic” bacterial pneumonia. IPF must be assumed always when, during auscultation of the lungs, the only additional phenomenon heard is crepitus, especially bilateral and spreading, without being replaced by moist rales, typical of the involvement of the walls of small bronchi in the inflammatory infiltration of the walls of small bronchi, which is characteristic of true bacterial pneumonia. Fever and other general symptoms of intoxication characteristic of pneumonia are usually absent in IPF: progressive respiratory failure comes to the fore in the clinical picture. In such patients, one should always first exclude interstitial lung diseases, in particular IPF, and long-term therapy with antibacterial drugs is harmful to them both from the point of view of a dramatic increase in the risk of adverse events, and due to the fact that more effective ones are not used. methods of pathogenetic therapy.
In general, today we can say that the use of high-resolution CT and surgical (including transthoracic) lung biopsy makes it possible to clearly confirm IPF in the majority of patients who actually suffer from this disease. The features of IPF that should be screened for by these diagnostic methods are defined in the 2011 guidelines (see table).
In contrast to diagnostic tactics for IPF, treatment tactics for this disease are still far from ideal, primarily because controlled clinical trials have not been able to confirm the effect of the therapeutic strategies most often used in routine clinical practice. The recommendations of the 2011 version recognize the use of glucocorticosteroids, most cytostatics (cyclophosphamide, azathioprine), cyclosporine, interferon-gamma-1, acetylcysteine and anticoagulants in monotherapy and in combinations in the treatment of IPF as inappropriate. Unfortunately, it is not yet possible to talk about the validity of the use of those drugs that are considered promising (etanercept, bosentan, which would be especially justified given the presence of pulmonary hypertension in the majority of patients with IPF), including the antifibrotic agent pirfenidone, which , according to individual clinical studies [26], has a positive effect on respiratory function and survival of patients with IPF. The discussion of sildenafil (its target of action is the same as that of the previously mentioned endothelin-1 antagonist bosentan) and imatinib mesylate in the 2011 recommendations did not lead to a statement of the advisability or inappropriateness of their use. Indications for starting long-term oxygen therapy are recommended to be determined individually. Lung transplantation is considered the preferred treatment for IPF, which has shown a positive effect on prognosis [27]. However, this section of the recommendations should not be accepted unconditionally, and in patients with IPF, especially with relatively early diagnosis, the tactics of immunosuppressive therapy can be active, including the use of glucocorticosteroids and cytostatics (for example, cyclophosphamide), including in ultra-high doses. Passively waiting for the development of indications for lung transplantation is unacceptable, including taking into account the low availability of this intervention in our country.
Of course, the 2011 version of the IPF recommendations is an important step forward towards developing consensus among experts regarding the management of this disease. Risk factors and approaches to diagnosing IPF are currently generally beyond doubt; The interdisciplinary approach to the management of IPF proposed by the expert committee is also key. The difficulties in determining therapeutic strategies and the lack of clear evidence of the effectiveness of most currently used drugs should stimulate new controlled clinical trials.
Diagnostic measures
Diagnosis of pulmonary fibrosis begins with collecting complaints, studying anamnesis and an objective examination of the patient. It is important to identify the presence of chronic diseases that can provoke the development of this pathology, as well as pay attention to typical clinical manifestations.
To confirm the diagnosis, the specialist prescribes a number of laboratory and instrumental studies, namely:
- General and biochemical blood tests.
- General urine analysis.
- Spirography. This research method allows you to evaluate the peak expiratory air flow rate, as well as the vital capacity of the lungs.
- Plain radiography of the chest organs, in at least two projections. In the image, the doctor reveals deformation and strengthening of the pulmonary pattern, especially in the lower sections.
- Computed and magnetic resonance imaging. Like the previous method, they can reveal structural changes in the lungs. However, with their help it is possible to conduct a more detailed and clear assessment of the pathological process.
How long do abnormal changes in the respiratory system last?
Post-Covid pathological changes persist in the lungs for a period of three to six to eight months. The duration is individual in each clinical case and depends on the state of the immune system, the presence of concomitant diseases, early or late treatment.
If even a slight manifestation of symptoms indicating the formation of pulmonary fibrosis appears, it is necessary to visit a doctor and undergo the recommended examination. The earlier treatment and pulmonary recovery are started, the lower the risk of complications.