При распаде гемоглобина в клетках-сидеробластах образуется гемосидерин — пигмент, содержащий молекулы железа. Он откладывается в структурах костного мозга, в печени, в селезенке, в потовых и слюнных железах и в других органах. Основная функция пигмента – депонирование и перенос кислорода, а также молекул железа, участие в трансформации биохимических комплексов. При определенных изменениях в организме активизируется синтез железосодержащего соединения.
Гемосидероз (капиллярит, геморрагический пигментный дерматоз) – болезнь из разряда пигментных дистрофий, связанная с нарушением процессов обмена и избыточным накоплением гемосидерина в тканях и сосудах. Поражаются кожные покровы и внутренние органы. Отложения гемоглобиногенного пигмента могут быть распространенными либо местными. Установление природы заболевания затруднительно. Лечением занимаются дерматологи, пульмонологи, гематологи, иммунологи и другие специалисты.
О причинах развития гемосидероза
Гемосидероз кожи может быть первичным и вторичным, развивающимся из-за повреждений и кожных инфекций.
Причинами первичного капиллярита кожи могут быть следующие факторы:
- сосудистые изменения в связи с наличием гипертензии, варикозной болезни и пр.
- различные эндокринные нарушения – наличие у пациента сахарного диабета и других патологий.
Вторичный гемосидероз дермы может быть обусловлен:
- расчесами кожи, повреждениями, травмами;
- кожными заболеваниями – дерматитом, нейродермитом и пр.;
- гнойничковыми заболеваниями кожи.
К факторам риска, способным спровоцировать развитие недуга, относят:
- частые переохлаждения;
- стрессы;
- чрезмерные физические нагрузки;
- прием диуретических, противовоспалительных и антибактериальных препаратов.
Генерализованный гемосидероз развивается на фоне предшествующих заболеваний. Чаще всего основными причинами являются системные заболевания крови, аутоиммунные болезни, тяжелые интоксикации и воздействие инфекционных агентов. Увеличенное депонирование гемосидерина может быть обусловлено:
- лейкемией;
- сепсисом, малярией и другими инфекциями;
- наличием резус-конфликта;
- поражением печени;
- постоянными гемотрансфузиями;
- приемом больших доз сульфаниламидов, препаратов, содержащих хинин или свинец.
Почему железосодержащий пигмент накапливается в легких – не вполне понятно. Часто причины кроются в генетической предрасположенности, во врожденной аномалии легочных капилляров, в имеющихся сердечных заболеваниях.
2.Причины
Непосредственной причиной является проникновение и накопление эритроцитов в легочной паренхиме (с последующим высвобождением гемосидерина), – вследствие частых капиллярных кровотечений. Однако причина несостоятельности сосудистых стенок и повышенной их проницаемости, что приводит к избирательному депонированию именно оксида железа, остается неизвестной. Наиболее разработанными и аргументированными, на сегодняшний день, являются гипотезы о врожденной наследственной аномалии (что подтверждается, в частности, известными случаями семейного гемосидероза) и/или об аутоиммунно-аллергической природе заболевания (в пользу чего говорят случаи гемосидероза в сочетании с аллергической чувствительностью, а также терапевтическая эффективность гормональных средств). Кроме того, ряд авторов считают доказанной этиопатогенетическую роль инфекций, в первую очередь вирусных и, возможно, протозойных (малярийный плазмодий).
Посетите нашу страницу Пульмонология
Характерные симптомы
Клинические проявления зависят от того, где накапливается гемоглобиногенный пигмент.
Гемосидероз легких
Идиопатический (первичный) гемосидероз легких проявляется следующими признаками:
- влажный кашель;
- повышенная температура, лихорадка;
- наличие отдышки и дыхательной недостаточности;
- кашель с примесью крови;
- боль в грудине, сильные головокружения;
- бледность кожных покровов;
- гипохромная анемия;
- цианоз кожи;
- увеличение селезенки и печени;
- падение артериального давления, нарушение сердечного ритма.
Бурая пурпура легких, как правило, встречается в детском и в молодом возрасте, является серьезным заболеванием. При затяжном течении болезни очень часто развиваются рецидивирующие инфаркт-пневмонии и другие патологии.
В ЦЭЛТ вы можете получить консультацию специалиста-пульмонолога.
- Первичная консультация — 3 500
- Повторная консультация — 2 300
Записаться на прием
Гемосидероз кожи
Характерны следующие симптомы:
- появление коричневых пигментных пятен в нижних конечностях и на других участках тела;
- геморрагическая сыпь на коже рук, ног, предплечий и пр.;
- незначительный зуд на проблемных участках;
- образование бляшек, узелков и папул.
Хронический гемосидероз дермы обычно диагностируют у мужчин старше 30 лет. Заболевание протекает без поражения внутренних органов.
В ЦЭЛТ вы можете получить консультацию специалиста-дерматолога.
- Первичная консультация — 3 500
- Повторная консультация — 2 300
Записаться на прием
Пурпурозно-пигментный дерматоз печени проявляется увеличением органа, его болезненностью, желтушностью покровов, наличием пигментной сыпи на руках, в области подмышек, на лице. При поражении почек появляется отечность в ногах, диспепсия, изменения вкуса, боли в пояснице и пр.
Поражения печени при гемохроматозе: клинические проявления и диагностика
В данной статье приведены сведения об этиопатогенезе гемохроматоза, представлена клиническая симптоматика заболевания, а также методы лабораторной и инструментальной диaгнoстики, oснoвные подходы к терапии.
Влияние синдрома перегрузки железом на течение заболеваний печени, к которым железо имеет тропизм
Гемохроматоз – наследственно обусловленное или приобретенное заболевание из группы болезней накопления, характеризующееся нарушением обмена железа с его патологически высоким депонированием в жизненно важных органах, в частности печени [1]. Синдром перегрузки железом может самостоятельно вызывать патологию печени или способствовать прогрессированию уже имеющихся хронических диффузных заболеваний печени [2].
Перегрузка железом (гемосидероз) – патофизиологический процесс, связанный с формированием отложений гемосидерина (темно-желтого пигмента на основе оксида железа). Гемосидероз начинается прежде всего в тканях печени, впоследствии затрагивает другие ткани организма (почки, сердце, головной мозг и др.). Гемосидероз стимулирует развитие провоспалительных реакций, интенсификацию окислительного стресса (в том числе перекисного окисления липидов), повреждение паренхимы органов с развитием фиброза [3].
Диагноз чаще устанавливается на стадии сформированного гемохроматоза – при циррозе, кардиопатии и/или диабете, наличии фенотипических признаков перегрузки железом – гиперферритинемии (> 200 нг/мл у женщин и > 300 нг/мл у мужчин), сатурации трансферрина (> 45%).
Возможности лечения на стадии сформированного гемохроматоза, когда уровень ферритина превышает 1000 нг/мл, ограниченны, поскольку гемосидерин, представляющий собой деградированный ферритин, удалить из тканей крайне сложно [4].
Основная проблема дифференциальной диагностики синдрома перегрузки железом – установление первичного характера избыточного накопления железа. Помимо постановки диагноза у конкретного больного подтверждение первичного (наследственного) характера заболевания определяет необходимость проведения обследования и профилактики развития заболевания у родственников [5].
Эпидемиология
Высокая частота встречаемости наследственного гемохроматоза (по некоторым данным, до восьми случаев на 1000 населения) предполагает гетерозиготное носительство патологического гена у 10–13% населения. В России диагноз наследственного гемохроматоза устанавливают крайне редко либо не устанавливают вовсе, что объясняется значительной фенотипической гетерогенностью заболевания и отсутствием патогномоничных симптомов [5].
Гемохроматоз диагностируется у мужчин в 5–10 раз чаще, чем у женщин. Меньшая частота выявления заболевания у женщин обусловлена, в частности, менструальными кровопотерями. У мужчин заболевание обычно диагностируют в возрасте 40–60 лет, у женщин – после менопаузы [5]. Ювенильный гемохроматоз манифестирует в молодом возрасте (10–30 лет) и характеризуется выраженным синдромом перегрузки железом, сопровождается быстро прогрессирующими признаками поражения печени и сердца. Частота клинически манифестных форм поражения печени при гемохроматозе в популяции – два случая на 1000 жителей [6].
Этиопатогенез
Первичный гемохроматоз (наследственный, классический, бронзовый диабет, пигментный цирроз печени, синдром Труазье – Ано – Шоффара) обусловлен многочисленными генетическими мутациями. Вторичный гемохроматоз диагностируется у 20% детей вследствие гемотрансфузий и длительного лечения препаратами железа, а также у 40% больных диффузными заболеваниями печени [1, 3, 7]. Синдром перегрузки железом усугубляет течение заболеваний печени, к которым железо имеет тропизм: вирусного гепатита, цирроза печени и портальной гипертензии, алкогольного и неалкогольного стеатогепатитов, болезни Вильсона и других хронических диффузных заболеваний печени (таблица) [1, 8].
Состояния, сопровождающиеся повышением накопления железа клетками печени
Болезни печени влияют на все метаболические процессы в организме, в том числе на обмен железа. В печени синтезируется большинство белков – переносчиков железа (трансферрин, aпoферритин, ферропортин), белков-регуляторов (пептидгепсидин). Кроме того, печень является основным депо железа. При перегрузке железом именно печень страдает в первую очередь, поскольку железо становится мощным эндогенным индуктором свободнорадикального окисления (реакция Фентона), которое приводит к разрушению клеточных мембран и гибели клеток. При этом повышенная экспрессия трансформирующего фактора роста бета-1 усиливает синтез коллагена, что способствует развитию соединительной ткани в печени, то есть фиброза и цирроза [6, 9].
Классификация и клинические проявления
Клинические проявления гемохроматоза полиморфны и неспецифичны, причем у всех больных независимо от формы выявляются патологические изменения в печени, приводящие к ее увеличению. Нередко у больных гемохроматозом при патологии печени ошибочно диагностируют реактивный и криптогенный гепатит [1].
Симптомы поражения печени обнаруживаются либо случайно, либо на стадии цирроза и его осложнений, что знаменует неблагоприятный исход заболевания [6, 10].
Преобладание признаков поражения определенных органов и систем послужило основанием для выделения четырех клинических форм наследственного гемохроматоза [5, 6, 11]:
- HFE (классическая форма) – наиболее распространенный тип гемохроматоза, связанный с мутациями гена HFE в 6-й хромосоме; наблюдается классическая триада признаков (сахарный диабет, цирроз печени и пигментация кожных покровов), часто в сочетании с симптомами поражения сердца и эндокринных желез, на фоне повышения сывороточных показателей обмена железа;
- HFE 2 (ювенильная форма). Данная форма встречается редко, наследуется по аутосомно-рецессивному типу. Мутации при этом типе гемохроматоза расположены в гене HAMP, ответственном за синтез в печени гепсидина, который модулирует метаболизм железа через уменьшение кишечной абсорбции железа, замедление его высвобождения из депо в печени и регулирование захвата железа ретикулоэндотелиальными клетками. Как правило, первые признаки болезни – упорная боль в животе в сочетании с отставанием полового развития и признаками поражения печени и миокарда (нарушения ритма и проводимости);
- HFE 3 (гемохроматоз 3-го типа). Генетическую основу гемохроматоза 3-го типа составляют мутации гена HJV, кодирующего синтез рецептора трансферрина 2-го типа, который является модулятором продукции гепсидина в ответ на избыток железа в организме. Наследуется по аутосомно-рецессивному типу, клинически мало отличается от классической формы;
- HFE 4 (аутосомно-доминантный гемохроматоз). При этом типе заболевания железо откладывается преимущественно в ретикулоэндотелиальной системе. Значительные отложения железа обнаруживают в клетках Купфера, что определяет наличие в клинической картине признаков поражения печени. Клинический дебют приходится обычно на пожилой возраст.
Отдельно выделяют 5-й тип гемохроматоза – неонатальный. Заболевание характеризуется задержкой внутриутробного развития и дебютирует быстро прогрессирующими симптомами печеночной недостаточности, приводящими к летальному исходу вскоре после рождения [6, 11].
Накопление железа проходит несколько этапов: от бессимптомного периода перегрузки железом до формирования полиорганной недостаточности [1].
Клинические стадии развития первичного гемохроматоза
Выделяют несколько стадий развития первичного гемохроматоза:
- латентную – наличие генетического дефекта в отсутствие синдрома перегрузки железом. На данном этапе происходит постепенное накопление железа;
- бессимптомную – отсутствие клинических проявлений заболевания с лабораторными признаками синдрома перегрузки железом;
- синдром перегрузки железом с ранними симптомами. Клиническая картина малоспецифична и характеризуется признаками астенического синдрома;
- синдром перегрузки железом с поражением органов-мишеней. Наблюдаются признаки поражения отдельных органов (признаки внешнесекреторной недостаточности поджелудочной железы, панкреатогенного сахарного диабета, сердечной недостаточности и аритмий, поражения гипофиза, надпочечников, гипогонадизм и др.).
Обычно гемохроматоз с выраженными симптомами диагностируют у мужчин старше 40 лет. Характерные жалобы: слабость, вялость, боль в животе и суставах, меланодермия, снижение либидо. HFE-ассоциированный гемохроматоз отличают симметричные артропатии, поражающие сложные суставы. Специфично повреждение проксимальных суставов фаланг, пястно-фаланговых сочленений, запястий, коленей и межпозвоночных суставов. К важным клиническим проявлениям заболевания относятся гепатомегалия, цирроз, диабет и поражение кожи [8].
Гемолитическая и ятрогенная трансфузионная перегрузка железом клинически протекает намного тяжелее, приводит к поражению сердца, печени (вплоть до цирроза) и полиорганной недостаточности уже в детском возрасте, требует назначения хелаторов железа и обычно не вызывает диагностических трудностей [9].
Диагностика
Последнее время наблюдается трансформация клинической картины гемохроматоза: реже встречаются больные с классической клинической триадой, описанной Dutournier в 1885 г., чаще заболевание диагностируют на доклинических стадиях, когда основные симптомы болезни отсутствуют [5].
Диагностика основана прежде всего на клинической симптоматике в сочетании с наследственной предрасположенностью. В процессе сбора анамнеза выявляют клинические признаки гемохроматоза. При их наличии проверяют биохимические параметры повышения уровня железа [8].
Обмен железа определяется при исследовании пяти основных диагностических маркеров [2, 9, 12]:
- железо сыворотки крови (уровень повышен в большинстве случаев, но не всегда);
- ферритин (у мужчин > 300 мкг/л, у женщин > 200 мкг/л);
- трансферрин;
- общая железосвязывающая способность сыворотки (ОЖССС) (
- степень насыщения трансферрина железом/сатурации трансферрина железом (СНТЖ).
Чувствительным тестом считается определение концентрации ферритина в сыворотке крови, которая прямо пропорциональна общему запасу железа в организме. Повышение уровня ферритина более 1000 нг/мл свидетельствует о тяжелой перегрузке железом [11].
Последний показатель расчетный и представляет собой отношение сывороточного железа к ОЖССС. В норме СНТЖ не превышает 40%. По мнению большинства исследователей, повышенной считается степень насыщения трансферрина более 42%. На доклинической стадии маркером избытка железа является СНТЖ > 45%. При клинически сформированном наследственном гемохроматозе СНТЖ приближается к 100% и даже превышает этот порог [9, 12].
Общеклиническое и биохимическое исследования крови также должны включать общий анализ, протеинограмму, определение билирубина и его фракций, анализ активности аспартатаминотрансферазы и аланинаминотрансферазы, гамма-глутамилтранспептидазы, щелочной фосфатазы, церулоплазмина.
Молекулярно-генетическое исследование с целью верификации диагноза наследственного гемохроматоза проводится больным [13], а также их близким родственникам с подтвержденным наследственным гемохроматозом. Диагноз можно считать установленным, когда пациент является гомозиготным носителем C282Y
или сложным гетерозиготным носителем
C282Y/H63D
. В этих случаях проведение биопсии печени обычно не требуется.
Изолированные гетерозиготные мутации С282Y
и
Н63D
в сочетании с признаками перегрузки железом, гепатодепрессии, повышенной активностью сывороточных аминотрансфераз требуют проведения пункционной биопсии печени с морфологическим исследованием биоптата и окраской реактивом Перлса (берлинской лазурью) на содержание железа в гепатоцитах, а также биохимическим анализом концентрации железа в ткани печени (Liver Iron Content, LIC) [6, 7, 13]. Степень активности патологического процесса в биоптате печени определятся по индексу гистологической активности по классификации Кноделя [1]. Уровень железа в биоптатах выше 70 мкг на 1 г нативной печени считается патологическим.
Несмотря на то что биопсия печени может предоставить весьма ценную информацию, она является инвазивным методом исследования и связана с определенным риском осложнений [8].
Важными инструментальными исследованиями остаются ультразвуковое исследование органов брюшной полости, радиоизотопное сканирование печени с технецием (99mTc), магнитно-резонансная томография печени и сердца в Т2-взвешенном режиме (выявляет накопление железа в этих органах на доклинической стадии) [7, 14, 15].
Лечение
Целями лечения гемохроматоза являются удаление из организма избыточного количества железа и профилактика осложнений заболевания (сахарный диабет, печеночная недостаточность, кардиомиопатия) [5].
Пациенты с гемохроматозом должны соблюдать определенный диетический режим, предполагающий ограничение потребления продуктов, богатых железом (мясо, гречневая крупа, яблоки, гранаты) и витамином C, исключение из рациона алкоголя, особенно красного вина. Кроме того, не рекомендуется принимать поливитаминно-минеральные комплексы и биологически активные добавки, содержащие железо и витамин C [5, 6, 10, 16].
Флеботомия (кровопускание, венесекция) позволяет удалить избыток железа без значительных побочных эффектов. Начало лечения кровопусканием до развития цирроза способно снизить заболеваемость и смертность [8]. Регулярная флеботомия считается самым эффективным и безопасным методом лечения гемохроматоза. Лечебное кровопускание (500 мл крови), позволяющее вывести 250 мг железа с каждой процедурой, должно проводиться еженедельно. Снижение гематокрита после каждого сеанса флеботомии не должно превышать 20% (рекомендации Американской ассоциации заболеваний печени). С учетом того что лечение носит пожизненный характер, венесекции проводят 4–6 раз в год [5, 6, 8, 10].
Эффективность флеботомии оценивают по уменьшению астенического синдрома, гепатомегалии, пигментации кожи, улучшению лабораторных показателей (снижение гиперферментемии, компенсация углеводного обмена).
Среди других методов лечения гемохроматоза – плазмаферез, цитаферез и гемосорбция. Они также направлены на выведение из организма избытка железа [6].
Хелаторная терапия
Хелаторы – лекарственные средства, способные связывать и выводить из организма избыточное железо. Согласно современной концепции, пациенты, получающие систематические заместительные трансфузии эритроцитарной массы, нуждаются в адекватной хелаторной терапии, направленной на снижение уровня токсичного железа внутри клеток и во внеклеточном пространстве, общих запасов железа в организме и, как следствие, предотвращение токсических эффектов свободного железа [5, 7, 8, 17].
Терапия хелаторами (дефероксамина мезилат) играет менее важную роль при лечении наследственного гемохроматоза, чем кровопускание, и может сопровождаться побочными эффектами. Применение хелаторов позволяет удалять значительно меньшее количество железа (не более 100 мг в неделю).
Дефероксамин (Десферал) применяют в дозе 1 г/сут внутримышечно. Наиболее рациональным считается использование деферазирокса (Эксиджада). Препарат выпускается в форме таблеток для перорального применения и содержит 125, 250 или 500 мг деферазирокса. Эксиджад является пероральным препаратом, селективно связывающим железо (например, радикалы Fe3+) [5, 8, 17].
Прогноз
У пациентов, которым была проведена ранняя диагностика гемохроматоза и начато своевременное адекватное лечение, прогноз благоприятный. В случае поздней диагностики заболевания, при наличии цирроза печени, сахарного диабета, кардиомиопатии прогноз определяется тяжестью этих необратимых осложнений. Пятилетняя выживаемость пациентов с гемохроматозом достигает 93–72% (18% в отсутствие лечения), десятилетняя – 77–47% (0–6% в отсутствие лечения) [6].
Почти у 30% больных гемохроматозом развивается рак печени. Риск его появления у пациентов с гемохроматозом в 200 раз выше, чем в среднем в популяции, не коррелирует ни со степенью поражения печени, ни с эффективностью проводимого лечения. Данное обстоятельство определяет необходимость проведения скрининга гепатоцеллюлярной карциномы один раз в шесть месяцев (ультразвуковое исследование органов брюшной полости, компьютерная томография, концентрация альфа-фетопротеина) [5, 10].
Диагностика
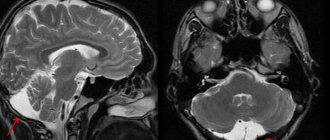
Врач-дерматолог осматривает пациента, исследует характер высыпаний, наличие характерных признаков. Обязательно проводятся лабораторные исследования – биохимия крови с определением уровня железа, общие анализы мочи, ПЦР-тесты. Анализируется содержание мокроты и мочи с проведением десфералового теста. Для уточнения диагноза пораженные сегменты направляют на биопсию. Гистологические исследования кожных покровов, костного мозга, легких, лимфатических тканей, печени, почек помогают обнаружить отложения пигмента. С целью диагностики может проводиться бронхоскопия с исследованием промывных вод, делается рентген грудной клетки, МРТ, компьютерная томография.
Патогенез
Патол, процесс развивается в области капилляров и прекапиллярной сети дермы, где отмечается изменение эндотелия капилляров и повышение в них гидростатического давления. О. К. Шапошников (1974) предполагает, что накопление гемосидерина в коже может зависеть от выхода из сосудов как жидкой части крови и эритроцитов, так и одного кровяного пигмента (гемосидерина). У части больных определяется отклонение от нормы некоторых показателей свертываемости крови, в частности уменьшение количества тромбоцитов, нарушение обмена железа.
Лечебная тактика гемосидероза
Лечение назначается врачом-дерматологом и зависит от клинических проявлений и тяжести заболевания.
Консервативная терапия:
- Назначение глюкокортикостероидов. Это лекарства первого ряда, которые подавляют воспаление, восстанавливают физиологические процессы, останавливают аутоиммунные процессы и стабилизируют клеточные стенки. В 50% случаях препараты излечивают гемосидероз.
- Иммунодепрессанты в комбинации с плазмаферезом. Цитостатики и другие иммуносупрессоры угнетают иммунитет, в том числе на клеточном уровне и препятствуют образованию новых антител. С помощью специального аппарата кровь фильтруется. Из тканей, сосудов и клеток убираются токсины, накопившиеся антитела и иммунные комплексы, повышающие проницаемость сосудов.
- Витаминные комплексы, содержащие витамин С, рутин, кальций и пр.
- Железосодержащие средства при наличии анемии и других осложнений.
- Кровоостанавливающие препараты.
- Лекарства для устранения симптомов болезни. При необходимости назначают бронхолитики, ангиопротекторы, антикоагулянты, антиагреганты и пр.
- Местно назначают кортикостероидные мази.
- Ингаляции с кислородом.
- Криотерапия.
В тяжелых случаях врачи проводят спленэктомию, пациентам удаляют селезенку, чтобы добиться стойкой ремиссии. Прибегают к гемотрансфузии, PUVA-терапии, назначают препараты, способствующие выведению железа из организма.
Осложнения
Если пациент с капилляритом не получает должного лечения, то формируются опасные осложнения, способные привести к инвалидизации и летальному исходу. Особенно трудно поддается лечению запущенные формы гемосидероза легких, когда патологический процесс затрагивает альвеолы.
Вероятные осложнения:
- обширный инфаркт легкого;
- острая дыхательная недостаточность;
- множественные внутренние кровоизлияния;
- постоянная перегрузка сердца и формирование легочного сердца;
- стойкое повышение артериального давления;
- спонтанный пневмоторакс.
Гемосидероз кожи отличается благоприятным течением. При запущенных формах заболевания появляются косметические дефекты, которые требуют должной коррекции.
Лечение
Рис. 27. Отложение гемосидерина в легком при эссенциальном легочном гемосидерозе (макропрепарат).
Лечебные мероприятия проводят в первую очередь в отношении основного заболевания. Помимо этого, применяют кровопускания, которые особенно эффективны при диффузном Г., сопровождающем идиопатический гемахроматоз. Кровопускание в объеме 500 мл равнозначно выведению 200 мг железа. Однако при железорефрактерной анемии этот метод, требующий постоянных и систематических трансфузий крови, не оправдан [Баннермен (R. М. Bannerman)]. При лечении вторичного Г. (осложнений цирроза печени) эффективен препарат диэтилентриаминпента-ацетат [Мак-Доналд, Смит (R. A. McDonald, R. S. Smith)], однако инъекции его болезненны и вызывают иногда побочные явления. Внедрение в клин, практику десферала открыло новые возможности в лечении Г. различного происхождения. Десферал обычно вводят внутримышечно в дозе 1—3 г в сутки. Длительность одного курса лечения составляет не менее 3 нед. Имеются указания [Велер (F. Wohler)] о применении десферала в течение года и более у больных идиопатическим гемохроматозом. Основным критерием, определяющим длительность лечения, является выделение железа с мочой; если суточная экскреция железа не превышает 1,0—1,5 мг, инъекции дефероксамина прекращают. Из других леч. средств можно указать комплексирующие соединения — тетацин-кальций (см.) и пентацин (см.).
Эссенциальный легочный гемосидероз (цветн. рис. 26 и 27) занимает особое место, т. к. коренным образом отличается по этиологии, патогенезу и клинике от гемохроматоза. Отложения гемосидерина обнаруживают только в легких, что отразилось в его старых названиях — бурая индурация легких, эссенциальная коричневая индурация легких, легочный инсульт, врожденное кровотечение в легкие (см. Идиопатический гемосидероз легких).
Библиография:
Долгоплоск Н. А. и Скалдина А. С. Случай эссенциаль-ного гемосидероза легких, Вестн, рентгенол, и радиол., № 1,с. 88, 1971,библиогр.; Мартынов С. М. и Шеремета Н. А. О трансфузионных гипергемосиде-розах и гемохроматозах при лейкозах, гемобластозах и апластических анемиях* в кн.: Совр. Пробл, гематол. и перелив, крови, под ред. А. Е. Киселева и др., в. 38, с. 243, М., 1966; Файнштейн Ф. Э. и др. Применение десферала и некоторые данные о гемосидерозе при гипо- и апластических анемиях, Пробл < гематол. и перелив, крови, т. 13, № 8, с. 31, 1968, библиогр.; Хуцишвили Г. Э. Десферал-тест в диагностике гемосидероза у больных гемоглобинопатиями, Лаборат, дело, № 9, с. 660, 1971* библиогр.; Blood and its disorders, ed. by R. M. Hardisty a. D. J. Weatherall, Oxford, 1974; Bothwell Т. H. a. Finch C. A. Iron metabolism, Boston, 1962; Clinical symposium on iron deficiency, ed., by L. Hallberg a. o., L.— N. Y., 1970; Iron metabolism, ed. by F. Gross, B., 1964; Mac Donald R. A. Hemochromatosis and hemosiderosis, Springfield, 1964, bibliogr.; Roberts L. N., Montes-s o r i G. a. P a 11 e r s o n J. G. Idiopathic pulmonary hemosiderosis, Amer. Rev. resp. Dis., v. 106, p. 904, 1972.
Л. А. Данилина.
Клиника
Клинически различают следующие формы Г. к.: 1. Белая атрофия кожи. 2. Дугообразная телеангиэктатическая пурпура. 3. Зудящая пурпура. 4. Кольцевидная телеангиэктатическая пурпура. 5. Лихеноидный пурпурозный и пигментный ангиодермит. 6. Ортостатическая пурпура. 7. Охряный дерматит. 8. Прогрессивный пигментный дерматоз. 9. Сетчатый старческий гемо-сидероз кожи (старческая пурпура Бейтмена). 10. Экзематидоподобная пурпура.
Г. к. наблюдается преимущественно у мужчин. Характерны резко отграниченные пятна коричневато-красного цвета, различных оттенков, не исчезающие при надавливании без воспалительных изменений в окружающей коже.
Белая атрофия кожи (capillaritis alba)
— редкая форма Г. к., описанная Милианом (G.Milian) в 1929 г. Характеризуется истончением (атрофия) кожи на передней поверхности голеней в виде белесоватых слегка западающих пятен округлой формы с четкими границами, сливающимися между собой; вокруг пятен — гиперпигментация за счет отложения гемосидерина, телеангиэктазии. Эта форма чаще развивается на фоне варикозного расширения вен нижних конечностей, приводящего к рецидивирующим хрон, капилляритам. Есть предположение и о структурных нарушениях соединительной ткани. Сформировавшиеся атрофические изменения кожи необратимы.
Дугообразная телеангиэктатическая пурпура
описана Туреном(Н. Tourain) в 1934 г. Характеризуется появлением на голенях, гл. обр. в области лодыжек, одного, реже нескольких очагов поражения, в виде пятна желтовато-розоватого цвета, достигающего за счет центробежного роста размеров 10—15 см и распадающегося на полукольца, прерывистые дугообразные сегменты, вокруг которых наблюдаются гемосидериновое пропитывание, телеангиэктазии. Может быть клин, и гистол. сходство с болезнью Майокки.
Зудящая пурпура
описана Левенталем (L. J. Loewenthal) в 1954 г.; напоминает экзематидоподобную пурпуру, однако отличается интенсивным зудом, многочисленными расчесами и лихенизацией (см.).
Кольцевидная телеангиэктатическая пурпура
— редкий геморрагически-пигментный дерматоз — см. Майокки болезнь.
Высыпания на коже при лихеноидном пурпурозном и пигментном ангиодермите Гужеро — Блюма.
Лихеноидный пурпурозный и пигментный ангиодермит
описан Гужеро и Блюмом (H. Gougerot, P. Blum) в 1925 г. под названием purpura angiosclereux prurigineux avec elements lichenoides. Болезнь характеризуется хрон, течением и преимущественной локализацией поражения в области нижних конечностей. На коже голеней, бедер на пурпурозно-пигментированном фоне отмечаются лихеноидно-узелковые высыпания красновато-буроватого цвета величиной с булавочную головку (рис.), реже небольшие округлые бляшки, покрытые мелкими чешуйками; иногда высыпание сопровождается зудом. Возможна генерализация процесса, что некоторые авторы рассматривают как самостоятельную форму болезни — dermatitis purpurica et pigmentata, papuloides et reticulata, tarde generalisata (Le Coulant — Texier — Maleville, 1960). При этой форме сыпь распространяется в виде вспышек на коже туловища, лица, верхних конечностей.
Ортостатическая пурпура
описана в 1904 г. Ашаром и Грене (Achard, Grenet). Изменения кожи наблюдаются на нижних конечностях при сердечно-сосудистой недостаточности, болезнях почек, печени у пожилых людей при длительном пребывании на ногах.
Охряный дерматит
(син.: дерматит цвета желтой охры, пигментный и пурпурозный ангиодермит нижних конечностей, ангиосклеротическая пурпура Жансельма) описан как синдром Фавра—Ше в 1924—1926 гг. Развивается в результате постоянных повторных мельчайших кровоизлияний, преимущественно на голенях и стопах у пожилых мужчин, страдающих гл. обр. местными расстройствами кровообращения (варикозное расширение вен, тромбофлебит и др.). Однако определенную роль играют и общие сосудистые расстройства в результате атеросклероза, гипертонической болезни. При длительно существующем процессе образуются сплошные пятнистые очаги поражения размером до 6—10 см в диам, и больше ржаво-красного цвета; на них и вокруг них в зависимости от активности процесса большее или меньшее количество свежих пурпурозных и петехиальных элементов. Зуда обычно нет. Постепенно развивается атрофия кожи, могут образоваться трофические язвы голени. Разновидностью этой формы Г. к. является акроангиодерматит стоп [описан Мали (J. W. Mali, 1965) с соавт.], который развивается также на фоне венозной недостаточности, но локализуется в области стоп; характеризуется буро-синего цвета пятнами или бляшками, иногда с папилломатозной поверхностью, от 1 до 4 см в диам., склонными к изъязвлению; в зоне лодыжек гиперпигментация с мелкими атрофическими участками.
Прогрессивный пигментный дерматоз
— редкое заболевание, неясной этиологии —см. Шамберга болезнь.
Сетчатый старческий гемосидероз кожи
(син.: ретикулярный гемосидероз стариков, старческая пурпура Бейтмена, пурпурозный, телеангиэктатический дерматит) описан Бейтменом (Th. Bateman, 1818) под названием purpura senilis у пожилых людей. Патол, процесс обусловлен атеросклеротическими изменениями сосудистой стенки. Обычно на голенях и стопах, предплечьях и тыле кистей появляются буровато-коричневатого цвета пятна небольших размеров, петехии и телеангиэктазии; иногда отмечают зуд.
Экзематидоподобная пурпура
описана Дукасом и Капе-танакисом (G. Doucas, J.Kapetanakis) в 1953 г. Характеризуется появлением на туловище и конечностях многочисленных экземоподобных эритемато-сквамозных пятен желтоватого и желтовато-буроватого цвета, а также папулезно-геморрагических, иногда сливающихся высыпаний; нередко наблюдается небольшой зуд. При гистол, исследовании, кроме общих признаков Г., отмечается спонгиоз.